Metallic Liver: C u Later!

Epidemiology of Wilson's Disease:
Wilson's disease (or hepatolenticular generation) is caused by an inborn error in copper metabolism by the copper-transporting gene, ATP7B. This gene expresses ATP7B protein, a protein that transports copper predominantly in the liver. If the gene is mutated, the protein it encodes is either absent, decreased in number, or functions improperly. It is inherited in an autosomal recessive fashion. Eventually, copper deposition within the liver leads to release of copper into the bloodstream and deposition into other critical organs including the cornea (leading to Kayser-Fleischer rings) and brain (neuropsychiatric symptoms).
The disease frequency has been estimated to be between 1 in 5,000 to 1 in 30,000 with a carrier frequency of up to 1 in 90; however studies out of Great Britain, Korea, and France have shown higher carrier frequencies of up to 1/25. Although the disease can present in older individuals (a frequently overlooked presentation), most cases present in patients younger than 40 years old. It is vital to recognize that 5-15% of symptomatic Wilson's patients may have normal or only slightly decreased ceruloplasmin, and up to 10-20% of heterozygotes may have reduced ceruloplasmin but are clinically asymptomatic.
Basic Pathophysiology:
Normal: ATP7B protein resides in the hepatocytes and signals 6 copper molecules to bind to apoceruloplasmin, forming ceruloplasmin. Ceruloplasmin (copper mover) transports copper and eliminates it through biliary system.
Abnormal: In Wilson's, there is a defective ATP7B protein and subsequent lack of elimination of copper into the biliary system, and hence, excess accumulation within the hepatocytes.
But why is ceruloplasmin low? If the ATP7B protein cannot signal copper molecules to bind to apoceruloplasmin, less ceruloplasmin is formed.
Clinical Manifestations:
- Hepatic: 40-50% of patients present with hepaticmanifestations as the initial manifestation including asymptomatic hepatomegaly, elevation of liver enzymes, to decompensated cirrhosis and acute fulminant hepatitis/liver failure. It can also mimic autoimmune hepatitis.
- Neurologic: Up to 40% of patients may also initially present with neurologic abnormalities as the primary manifestation including tremor, dysarthria, slurred speech, dystonia, cerebellar dysfunction with ataxia and less frequently, seizures, tics, myoclonus, chorea.
- Psychiatricsymptoms are less common (mood disorders, psychosis)
- Ophthalmological: German Ophthalmologists Dr. Kayser and Dr. Fleischer described pigmented corneal rings 10 years prior to the discovery of Wilson's disease by Dr. Wilson (Great Britain) in 1912, but the connection between KF-rings and Wilson's was formally made by Dr. Fleischer in his 1912 publication.
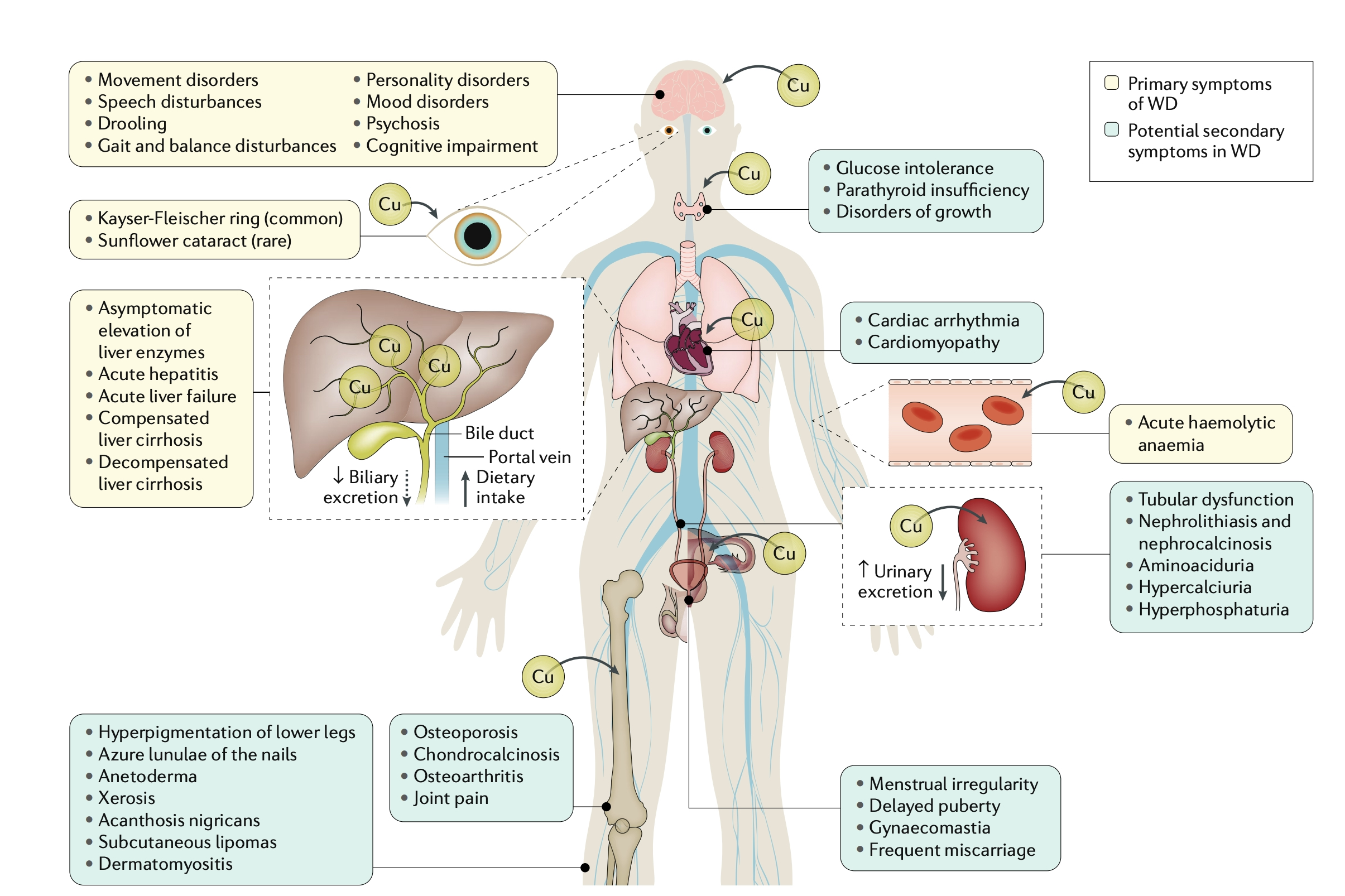
Image taken from: Członkowska, A et al. Wilson disease. Nat Rev Dis Primers 4, 21 (2018).
Diagnostic Algorithm
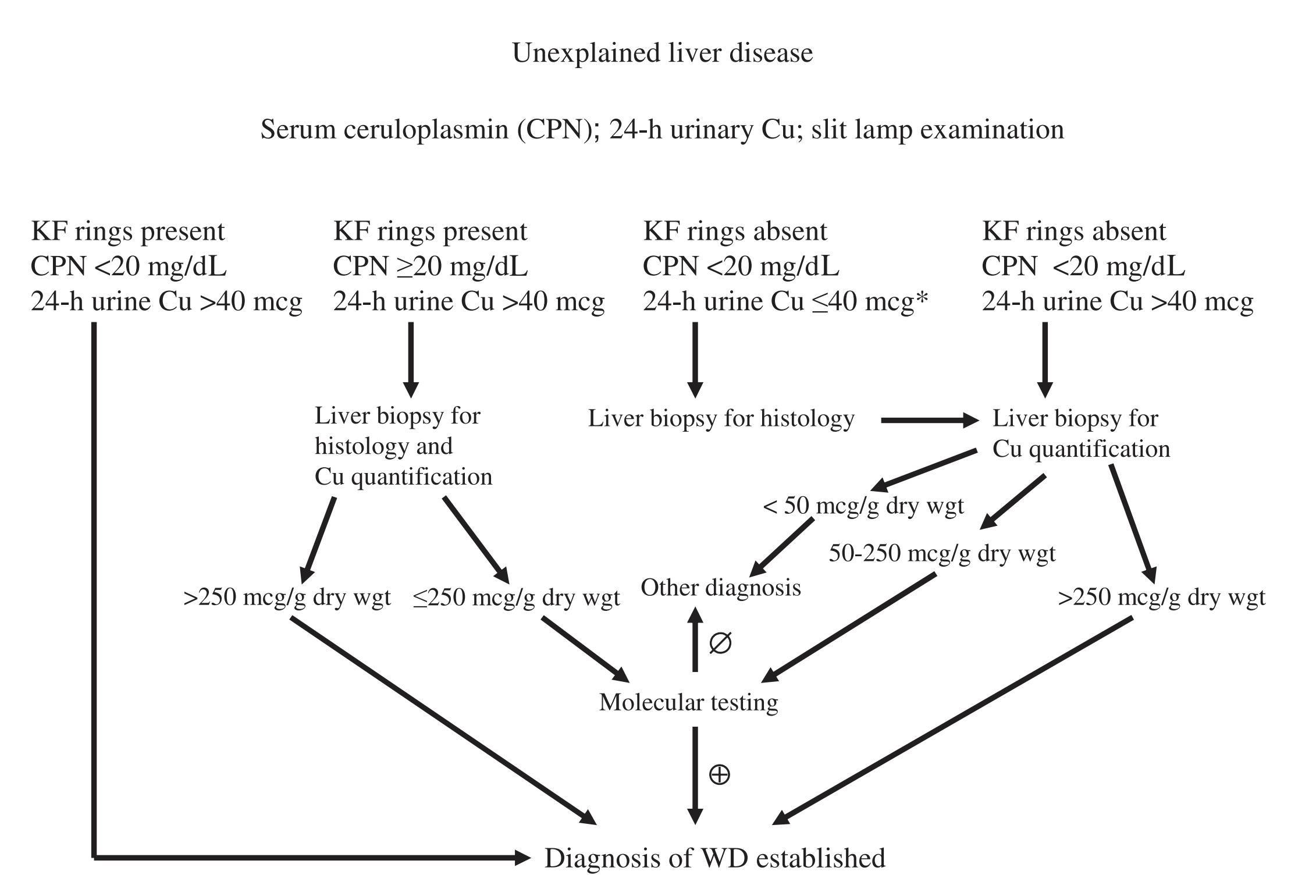
Image taken from: Roberts EA et al. Diagnosis and treatment of Wilson disease: an update. Hepatology 2008; 47: 2089– 2111
Other important points:
- The most sensitive and accurate test is liver biopsy
(Elevations greater than 250ug/g of dry tissue) typically present for definitive diagnosis.
- A serum copper level may be low, normal, or elevated in patient’s with Wilson’s disease and therefore may not helpful in supporting your diagnosis.
- If there are neurological symptoms, neurology consultation and radiological imaging (MRI) should be considered prior to treatment.
- Other populations that benefit from work-up for Wilson’s according to AASLD guidelines
- Pediatric age bracket with clinical picture of autoimmune hepatitis
- Adults with atypical autoimmune hepatitis who do not respond well to corticosteroid therapy
- Should be within the differential diagnosis in patients with NAFLD or NASH
- Suspected in patients with acute liver failure with coombs-negative intravascular hemolysis, elevated aminotransferases, or low alkaline phosphatase (ALK) with ALK/Total Bilirubin ratio <2
- All first-degree relatives of newly diagnosed Wilson’s Disease must be screened.
- Chelators: (mechanism: induces cupruria)
- D-Penicillamine
- Most side effects (e.g. bone marrow toxicity, nephrotoxicity, neutropenia, thrombocytopenia, up to 30% of patients will need to discontinue)
- Trientine
- Fairly well-tolerated with few side effects
- Zinc: (mechanism: prevent copper absorption) - Well-tolerated
- First-line therapy for asymptomatic or pre-symptomatic patients
- D-Penicillamine
How to administer: Treatments above should not be administered with food
Adequacy and compliance of treatment: Measured by monitored 24-hour urinary copper excretion
- Diet: Avoid shellfish, nuts, chocolate, mushrooms, organ meats
- Environment:
- Well water or water brought through homes in copper pipes must be checked for copper content.
- Avoid cooking or storing food in copper cookware/containers.
So who and when do we treat?
Asymptomatic: Zinc or low dose D-Penicillamine
Maintenance Therapy: Zinc or low-dose chelator after adequate initial treatment with high-dose chelator
Decompensated Cirrhosis:Chelator (either D-Penicillamine or Trientine) + Zinc combination therapy (Note: must be dosed 5-6 hours apart to avoid chelation of Zinc)
Acute Liver Failure: Liver Transplantation (Until transplantation, plasmapheresis, hemofiltration, exchange transfusion, or dialysis should be performed to protect kidneys from tubular damage).
Prognosis indicators:
Nazer Prognostic Index (1986): Helps determine probability of survival without liver transplant
- 34 patients with Wilson’s included in study (27 symptomatic index cases, 7 asymptomatic patients)
- Scores great than 7 died without transplantation
- Scores less than/equal to 6 survived

Newer prognostic tools including New Wilson Index, CTP score, and MELD are also used in evaluation.
Liver Transplantation
Indications in Wilson’s Disease
- Presentation consistent with acute liver failure
- Decompensated cirrhosis unresponsive to medical therapy
Transplant Takeaways:
- 1-year overall survival is >80% and those who survive the first year tend do well long-term
- Living donor transplantation can be considered and performed
- Note: successful when donor family member is heterozygous for Wilson’s disease
- Some patients with neuropsychiatric manifestations, may have improvement post-transplant
Not recommended for primary neurologic Wilson’s disease