In the Beginning: Neonatal Screening and Management of Wilson Disease

Introduction
Wilson's disease (WD) is an inherited disorder affecting copper metabolism, resulting from homozygous or compound heterozygous mutations in the ATP7B gene. This gene encodes ATP7B, a transmembrane copper-transporting ATPase responsible for copper excretion into bile and delivery for ceruloplasmin synthesis. In WD, defective ATP7B function leads to copper overload in hepatocytes, causing liver pathology. Excess copper, not bound to ceruloplasmin, is released into circulation, leading to pathological accumulation in various tissues, particularly the brain, resulting in neurological and psychiatric symptoms. Wilson's disease typically manifests between ages 5 and 35. In pediatric cases, hepatic symptoms, including incidental elevation in liver enzymes, hepatomegaly, and jaundice, commonly present, with the potential progression to liver failure. Conversely, adults predominantly manifest psychiatric symptoms, encompassing personality changes, mood swings, and cognitive impairment. Neurological manifestations, such as tremors and dystonia, are consistently observed across age groups. WD is a rare genetic disorder, with an approximate incidence of 1 case per ~30,000 individuals. Recent molecular studies indicate an increased prevalence. Early diagnosis and precise treatment are imperative for successful management, as untreated WD universally results in fatality. The diagnosis and management in adults and adolescents have been discussed in "Metallic Liver: C u Later! | AASLD," this post focuses on the role of neonatal screening for Wilson disease.
As per the AASLD guidelines, screening of first-degree relatives of an individual affected by Wilson disease is recommended given the variability in the age and mode of presentations. Screening offspring of parents with known pathogenic ATP7B mutations is also advised. The widespread availability of genetic testing has led to the identification of asymptomatic infants. Managing such cases poses challenges due to the scarcity of medical literature.
Genetics of Wilson disease
Wilson's disease (WD) is a hereditary disorder stemming from mutations in the ATP7B gene on chromosome 13. The ATP7B gene exhibits over 700 mutations, primarily consisting of missense or nonsense mutations. Patients can be homozygous for one mutation or carry two different mutations as compound heterozygotes. A clear genotype-phenotype does not exist in Wilson disease. The lack of a distinct genotype-phenotype correlation is likely attributed to intricate interactions involving genetics, epigenetics, and the environment.
Copper metabolism
Copper is an essential metal crucial for the function of numerous metalloproteins. Enterocytes in the duodenum and proximal small intestine absorb a portion of dietary copper (average 2–5 mg/day) to meet the recommended intake of 0.9 mg/day. The absorbed copper, mainly associated with albumin, is transported in the portal circulation and efficiently cleared by the liver. Hepatocytes utilize copper for metabolic processes, integrate it into nascent ceruloplasmin, and eliminate excess copper into bile. The primary route for excreting excess copper is through bile into feces, with only a minor amount being renally excreted. Impaired biliary copper excretion results in hepatic copper retention.
Fetal copper transportation involves ATP7A and ATP7B transporters, both crucial for copper homeostasis in the placenta. ATP7A facilitates copper import to the fetus from maternal circulation, while ATP7B allows copper efflux back to the mother. In the third trimester, fetal copper needs increase, diminishing the importance of efflux. In infants with defective placental ATP7B, it's uncertain if total body copper is appropriate at birth.
‘High' copper foods, like organ meat, shellfish, mushrooms, chocolate, and nuts, are rarely consumed by infants, making dietary exclusion uncomplicated. Infant formula and baby foods, however, merit attention for their substantial copper content.
Presentations of infantile Wilson disease:
There are only a few documented cases of infantile Wilson disease. Most patients are detected through genetic testing as part of screening programs. However, a small number of symptomatic cases have been observed. In these instances, the primary presentation was a silent elevation of associated liver enzymes.
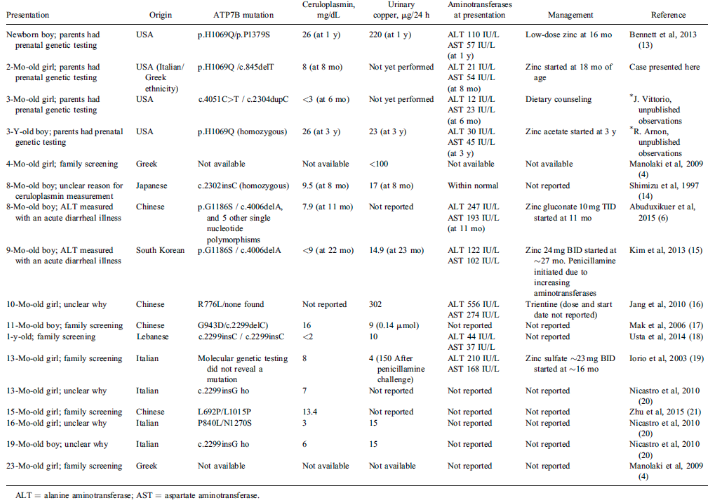
Table source: Valentino, P. L. et al. Management of Wilson disease diagnosed in infancy. Journal of Pediatric Gastroenterology and Nutrition 70, 547–554 (2020).
Management:
Currently there is no consensus regarding the management of infantile Wilson disease. The treatment can be divided into a surveillance phase and a treatment phase.
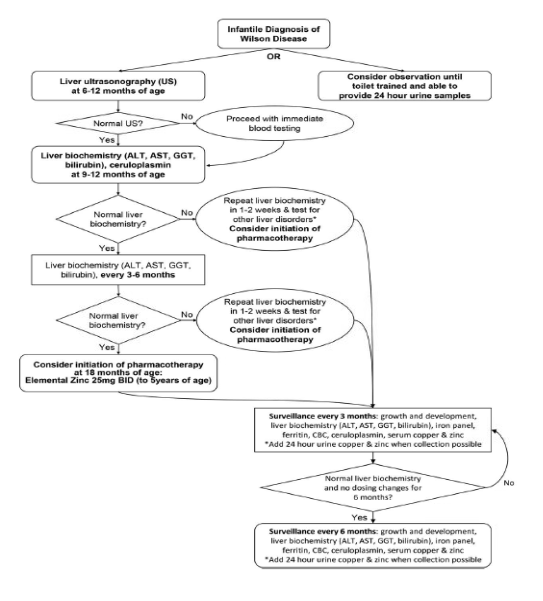
Initiating surveillance in infants requires careful consideration, recognizing the challenges in obtaining blood samples. Assessing the liver biochemical profile of patients with genetically confirmed WD at 9-12 months, followed by serial assessment every 3-6 months, is appropriate. A significant increase in liver-associated enzymes merits exploring alternative etiologies. Performing a liver ultrasound between 6-12 months aids in establishing a baseline and screening for potential changes in liver echogenicity.
Treatment:
Initiating drug therapy for infantile Wilson disease lacks strong evidence. Based on our understanding of the pathophysiology of Wilson's disease (WD), it is reasonable to infer that infants with WD haven't had the chance to accumulate substantial amounts of hepatic copper. Given this restricted copper content, zinc emerges as the preferred therapeutic intervention. Additionally, due to the severe side effects associated with penicillamine and trientine, their usage in this patient population has not been described in the literature. Using oral zinc, a well-tolerated treatment, requires close monitoring for copper deficiency. The recommended dose for infants is 25mg twice daily. According to the limited available literature, zinc therapy is generally well tolerated, with infrequent adverse effects documented. These adverse effects primarily consist of gastrointestinal discomfort and diarrhea. Delaying treatment until the emergence of abnormal liver biochemistry or until 24-hour urinary copper collection is developmentally possible may be considered. Individualized care is recommended for each patient.
Proposed schema for the management of infantile Wilson disease
Future directions: Universal screening for Wilson disease
The incorporation of a disease into the U.S. newborn screening panel necessitates an exhaustive assessment, encompassing factors such as disease severity, treatment efficacy, testing reliability, prevalence, disease natural history, healthcare consensus, and ethical considerations. Wilson disease notably aligns with and fulfills these rigorous criteria on a global scale. Numerous studies have been conducted to identify an optimal serum biomarker candidate suitable for assessment through the dried blood spot technique. A study identified ATP7B as a potential marker. A recent investigation further examined the efficacy of ceruloplasmin as a potential biomarker, yielding promising results. These studies support the idea of newborn screening for Wilson disease. However, the absence of cost-effectiveness studies on screening for Wilson's disease, combined with the benign nature of the disease in the immediate neonatal period, raises doubts about the utility of this approach. Lastly, advancements in genetic diagnosis and emerging therapies for genetic disorders, the landscape of the newborn screening panel may witness the addition of more diseases.