Pathology Pearls: The 3 C's - Congenital Hepatic Fibrosis, Caroli disease & Caroli Syndrome

Brief Case Presentation
A 12-year-old female presents with an acute onset of fever and abdominal pain. On the physical examination, the patient’s sclerae and mucous membranes show evidence of jaundice. In addition to leukocytosis, the results from her liver chemistry tests are seen below:
| Lab Test | Result |
| AST | 106 U/L (8 – 37 U/L) |
| ALT | 63 U/L (8 – 35 U/L) |
| Total Bilirubin | 2.6 mg/dL (0.1 – 1.0 mg/dL) |
| Conjugated Bilirubin | 1.6 mg/dL (0.0-0.3 mg/dL) |
| Unconjugated Bilirubin | 1.1 (0.1-1.0 mg/dL) |
| Alk. Phos. | 404 U/L (50-150 U/L) |
The ultrasound revealed increased hepatic echogenicity with multiple bilobar hypoechoic hepatic areas in the porta hepatis, consistent with cysts (Fig. 1). There were no abnormalities of the common bile duct. The ultrasound also showed evidence of portal hypertension, including biphasic portal vein flow, splenomegaly and small amount of ascites. The kidneys were bilaterally enlarged with poor corticomedullary differentiation and multiple small cysts.
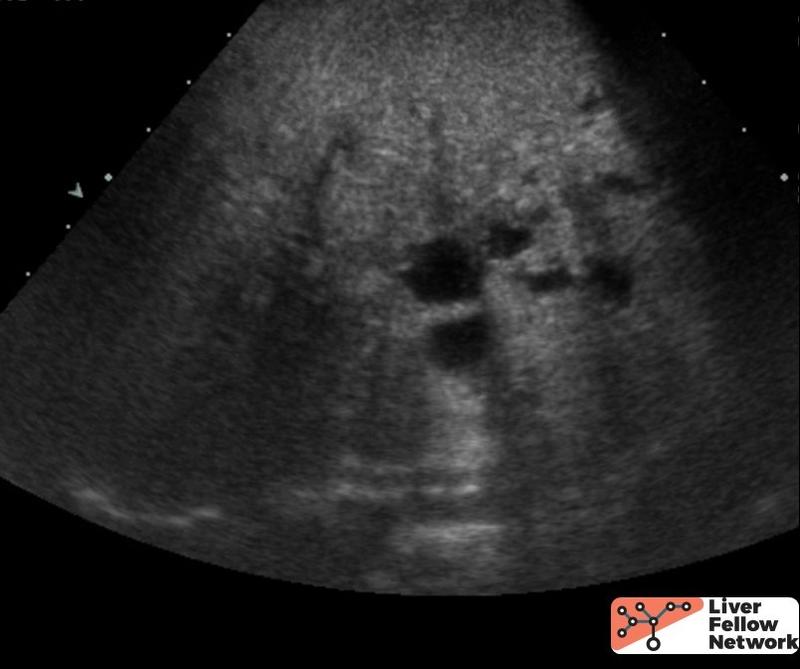
The patient was treated for acute cholangitis and underwent extensive investigations, including genetic testing which identified a pathogenic variant of PKHD1 gene. She continued to have recurrent bouts of ascending cholangitis and ultimately underwent a liver transplant.
Liver Transplant - Macroscopic and Microscopic Findings
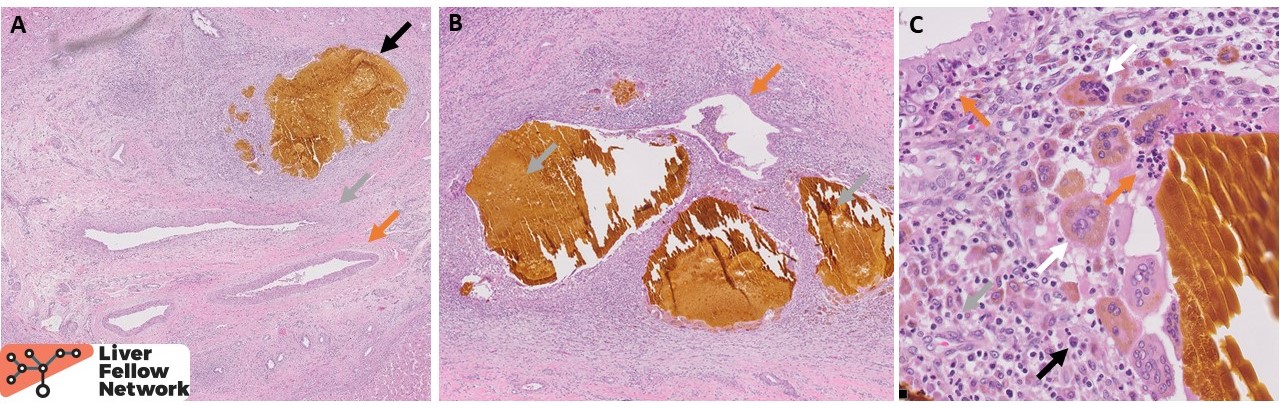
The macroscopic explant specimen revealed a diffusely nodular, bile-tinged liver parenchyma with scattered dilated bile ducts filled with bile sludge (Fig. 2). A section from one such area shows dilated large intrahepatic bile ducts surrounded by acute and chronic inflammation (Fig. 3). Some of the ducts are ruptured, expulsing large lakes of bile into the parenchyma, inciting a prominent multinucleated giant cell reaction.

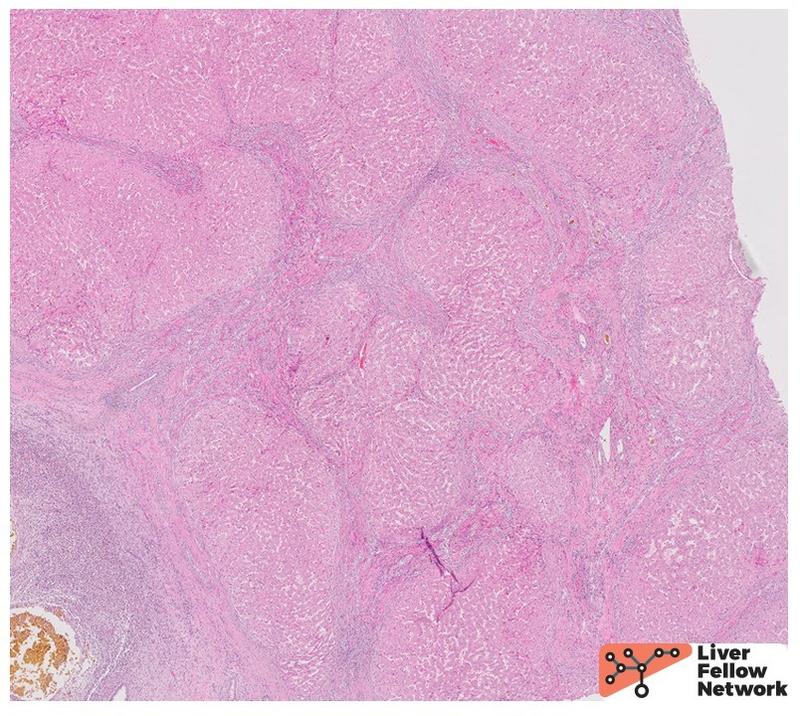
The smaller ducts also show histologic abnormalities, with ectatic dilation and anastomosing pattern and occasional spillage into the periseptal hepatic parenchyma (Fig. 4A). Some of the ducts are seen circling around mesenchymal cores, consistent with ductal plate malformation (Fig. 4B). The background liver shows irregular fibrous bands separating the liver parenchyma into undulating islands (Fig. 5).
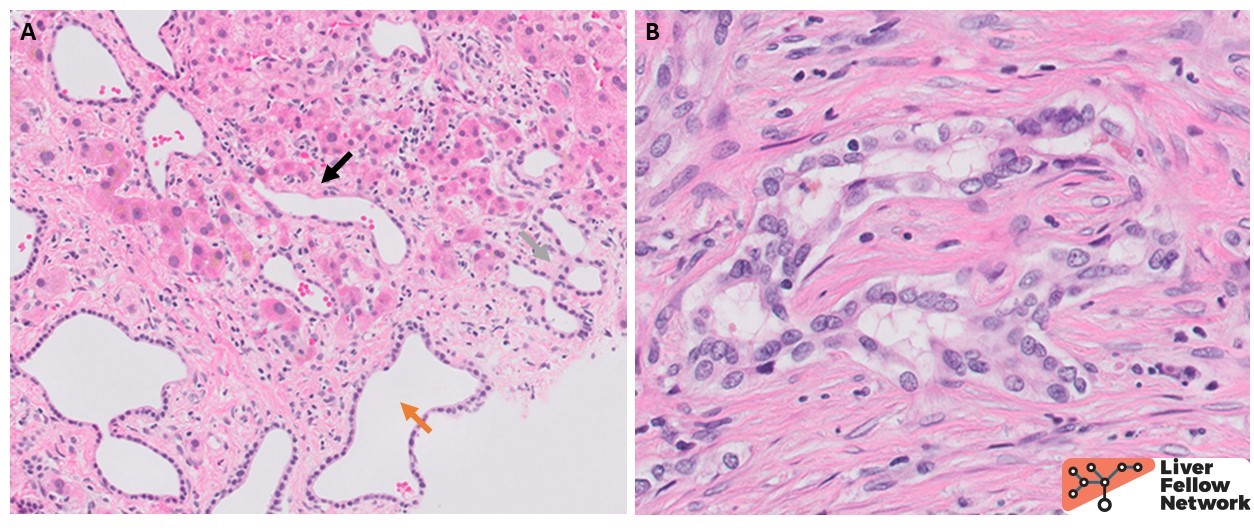
In light of the clinical history and the presence of PKHD1 variant, the dilated large intrahepatic ducts and the characteristic pattern of fibrosis are diagnostic of Caroli syndrome.
Congenital Hepatic Fibrosis, Caroli Disease & Caroli Syndrome
Definitions
Congenital hepatic fibrosis (CHF), Caroli disease and Caroli syndrome are three congenital conditions of abnormal embryonal biliary system remodeling, associated with a spectrum of macroscopic and microscopic cystic lesions of the liver. In CHF, ductal abnormalities are limited to the small interlobular bile ducts and are associated with progressive hepatic fibrosis. On the other hand, Caroli disease is characterized by segmental dilations of the large intrahepatic bile ducts. When the two coincide, Caroli syndrome is diagnosed.
Associated Conditions
All of these conditions are most commonly associated with a germline PKHD1 mutation and co-occur with kidney abnormalities, and as such are often considered to be part of the spectrum of autosomal recessive polycystic kidney disorder (ARPKD). PKHD1 encodes fibrocystin found in cilia of cholangiocytes in addition to renal collecting duct epithelium, and its loss is believed to abrogate the normal ductal plate development in liver. Less frequently, CHF, Caroli disease or Caroli syndrome arise in the context of other hepatorenal fibrocystic disorders such as nephronophthisis, medullary cystic kidney disease, Joubert syndrome, Meckel-Gruber syndrome, Bardet-Biedl syndrome, COACH syndrome (cerebellar vermis hypo/aplasia, oligophrenia, congenital ataxia, ocular coloboma, and hepatic fibrosis), or tuberous sclerosis. In rare instances, they have been documented in association with autosomal dominant polycystic kidney disease (ADPKD) or without extrahepatic abnormalities.
Clinical Characteristics
CHF affects approximately 1 in 20,000 live births while Caroli syndrome is seen in 1 in 100,000 live births and Caroli disease in 1 in 1,000,000 live births. Most patients become symptomatic in childhood or early adulthood. The clinical manifestations vary, in part due to genetic heterogeneity in the underlying gene alterations, associated extrahepatic disease and inherent features of the disease. The portal fibrosis and developmental abnormalities in CHF lead to narrowing of portal veins and secondary non-cirrhotic portal hypertension, so the affected patients commonly present with secondary sequelae, such as variceal bleeding, hepatosplenomegaly, or ascites. On the other hand, the main abnormality in Caroli disease is the ectatic large ducts which are prone to superimposed infections, and so the affected patients usually come to clinical attention due to ascending cholangitis or sepsis. In Caroli syndrome, both can occur as seen in this patient. Not uncommonly, extrahepatic disease, particularly renal or pulmonary, will be the first presentation that will trigger the genetic testing. The patients in all three conditions are usually managed supportively unless complications from portal hypertension or recurrent ascending cholangitis oblige surgical interventions, including partial hepatectomy for unilobar disease or liver transplant.
Diagnostic Modalities
Imaging in CHF can highlight the sequelae of portal hypertension and changes in liver texture. However, the definite diagnosis of CHF requires a histologic confirmation. In contrast, radiologic documentation of cystically dilated large proximal intrahepatic bile ducts, especially in the presence of the central dot sign (small focus of contrast enhancement within the duct which correlates to the fibrous cords containing portal vessels within the cystic cavities) and a normal common bile duct is usually sufficient for diagnosis of Caroli disease in the appropriate clinical context, while a biopsy can carry increased risk of complications in the setting of ectatic ducts.
Pathologic Features
The macroscopic examination of liver in CHF patients reveals hepatomegaly and a fine reticular fibrosis but no grossly appreciable cysts. Caroli disease, on the other hand, is characterized by visible cystically dilated ducts variably filled with bile or bilirubin calculi and occasionally traversed by fibrous bridges with embedded portal veins.
The unifying histologic feature in all three conditions is the presence of ductal plate abnormalities which can vary from small anastomosing ducts circling around a fibrous core to anastomosing, dilated biliary channels, occasionally herniating in the adjacent hepatocellular parenchyma (Fig. 4). Unlike in ADPKD, these channels are connected to the biliary system and often contain bile. In addition to ductal plate abnormalities, CHF shows diffuse periportal fibrosis forming irregular fibrous bands (Fig. 5). Increasing thickness of the latter leads to narrowing of portal veins and hepatic venules which explains the non-cirrhotic portal hypertension. The fibrosis separates the hepatocytes into irregular islands reminiscent of an unfinished jigsaw puzzle. However, there are no actual regenerative nodules of hepatocytes in the true sense of the term cirrhosis – as such, CHF is a bit of a misnomer. In absence of superimposed infection, the inflammation is minimal in CHF. On the other hand, Caroli disease is characterized by dilated large intrahepatic ducts with thickened, variably fibrotic walls surrounded by severe chronic inflammation with lymphocytes, occasional lymphoid follicles and plasma cells (Fig. 3). If superimposed cholangitis is present, acute inflammation can be seen in the form of neutrophils infiltrating ducts and ductules as well as the surrounding connective tissue, as seen in the current case. Repeated inflammation may lead to ulceration and loss of ductal epithelium. Rarely, epithelial dysplasia can be seen. Most importantly, the histologic evaluation may reveal the most dreadful complication of Caroli disease, a cholangiocarcinoma.
Comparison with ADPKD
As seen in the previous Pathology Pearls vignette, the patients with ADPKD tend to present as older adults, primarily with renal complications. The condition is caused by mutations in PKD1 and PKD2 genes which also lead to primary cilia dysfunction but without ductal plate abnormalities. The intrahepatic cysts are not connected with the biliary system and are not associated with significant inflammation. Numerous bile duct hamartomas (Von Meyenberg complexes) are a histologic clue.
References
- Burt, MacSween's Pathology of the Liver: Seventh Edition. 2017.
- Suchy FJ. (2023). Caroli disease. UptoDate. Available from https://www.uptodate.com/contents/caroli-disease#H855304549
- Acevedo E, Laínez SS, Cáceres Cano PA, Vivar D. Caroli's Syndrome: An Early Presentation. Cureus. 2020;12(10):e11029.
- Hasbaoui BE, Rifai Z, Saghir S, et al. Congenital hepatic fibrosis: case report and review of literature. Pan Afr Med J. 2021;38:188.
- Umar J, Kudaravalli P, John S. Caroli Disease. In: StatPearls. Treasure Island (FL): StatPearls Publishing; August 1, 2022.
- Hartung MP. (2022). Caroli disease. Radiopaedia. Available from https://radiopaedia.org/articles/caroli-disease-1?lang=us