Pathology Pearls: Hepatobiliary Manifestations of Cystic Fibrosis

Brief Case Presentation
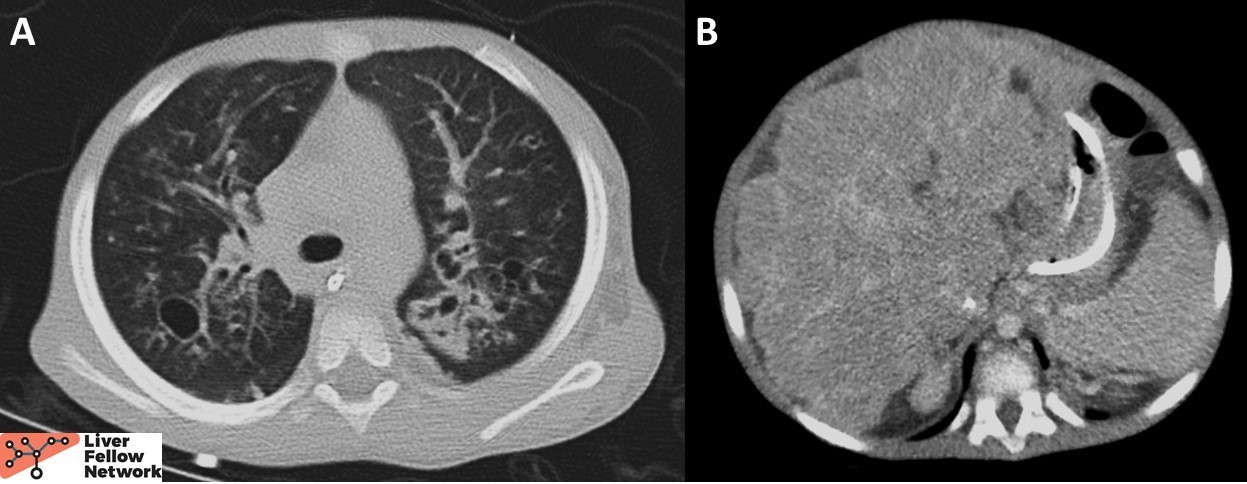
A 3-year-old male presented with respiratory distress and worsening chronic abdominal distension. CT was notable for bilateral bronchiectasis with bronchial wall thickening and cavity formation, as well as nodular liver morphology (Figure 1). Respiratory PCR panel came back positive for rhinovirus. Viral hepatitis panel, autoimmune hepatitis serologies and the metabolic disorder panel were negative. Alpha-1-antitrypsin phenotyping revealed PI*MM phenotype.
|
Lab Value |
Result |
|
AST |
131 U/L (8-37 U/L) |
|
ALT |
100 U/L (8-35 U/L) |
|
Total Bilirubin |
4.7 mg/dL (0.1-1.0 mg/dL) |
|
ALP |
113 U/L (100-390 U/L) |
|
INR |
3.3 (0.9-1.1) |
|
|
Figure 1. A. CT chest revealed bronchiectasis with bronchial wall thickening and cavity formation. B. CT abdomen demonstrated a nodular, markedly heterogeneous liver. |
A sweat chloride test was performed and revealed a chloride concentration of 80 mmol/L (normal <30 mmol/L). Subsequent genetic testing confirmed two mutated CFTR alleles. His hepatobiliary disease progressed over the course of the year, eventually requiring a liver transplant.
Liver transplant
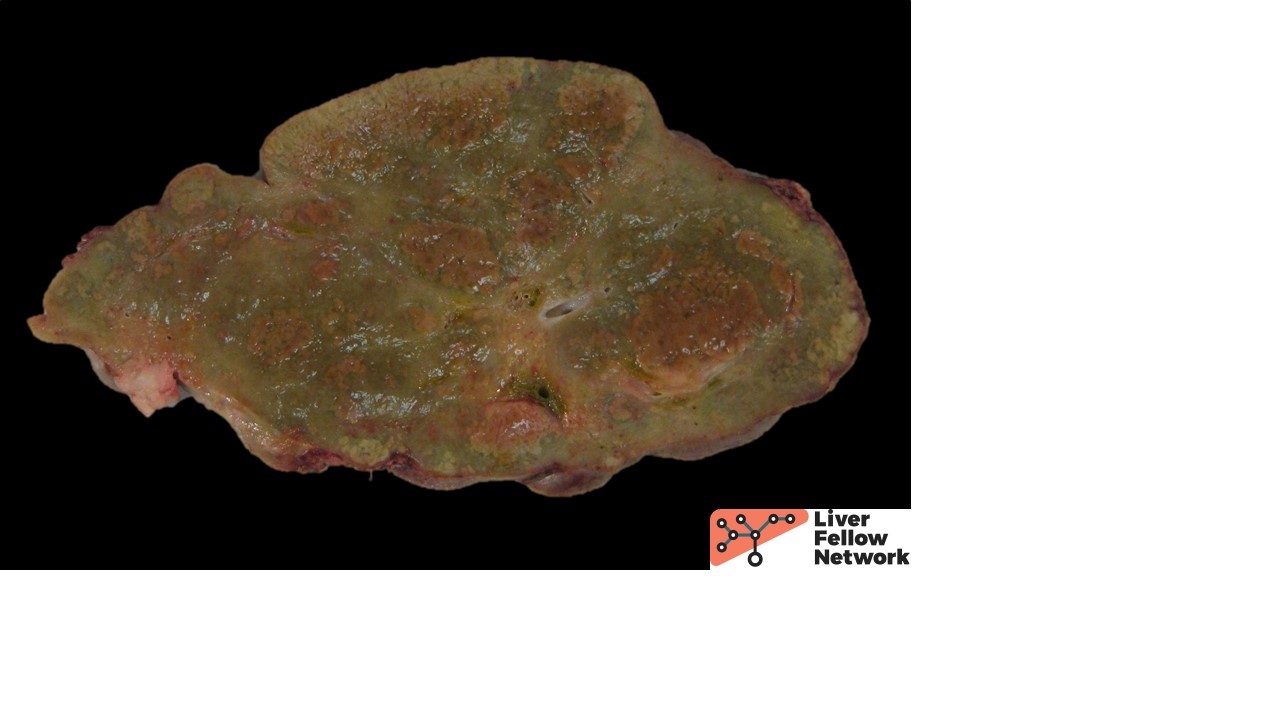
The macroscopic explant specimen revealed a tan-green liver parenchyma with occasional large nodules surrounded by irregular, depressed, fibrous bands (Figure 2).
|
|
Figure 2. This cut section from the nodular parenchyma shows irregular bands separating variably-sized hepatocellular nodules. |
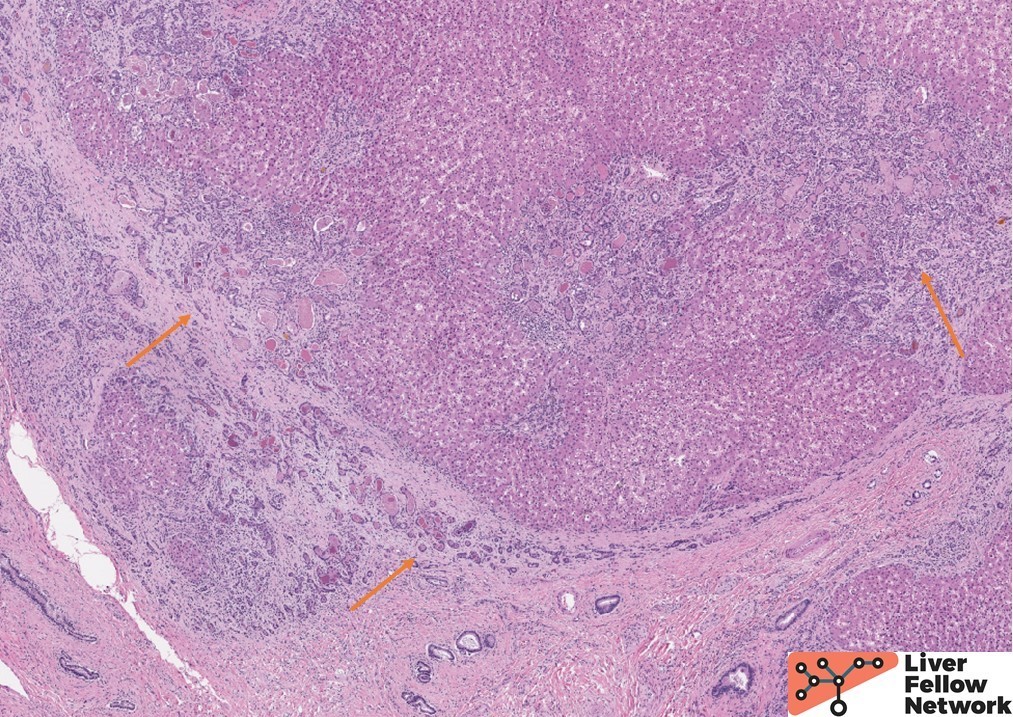
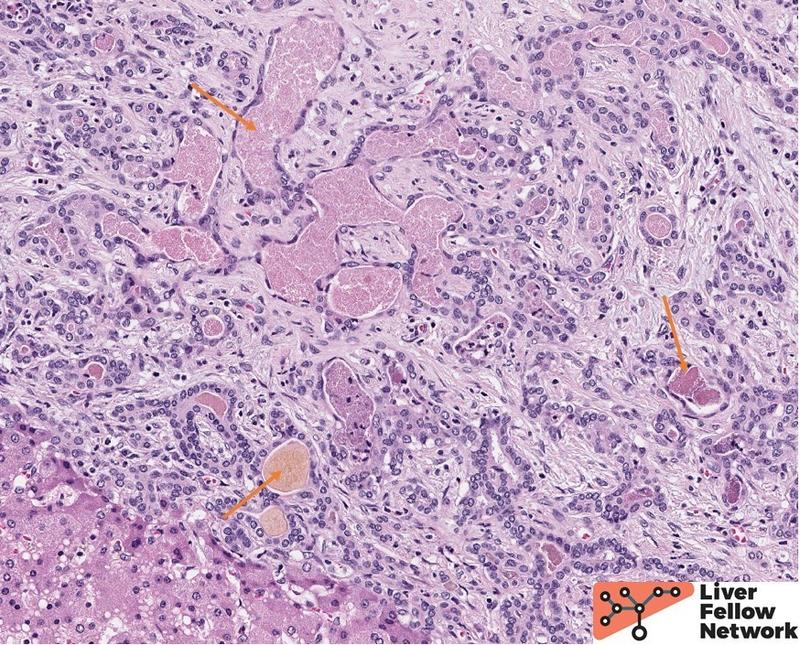
A histologic section from one such area shows irregular islets of hepatocytes surrounded by fibrosis and prominent ductular reaction (Figure 3). The ductules are filled with dense, pink and occasionally orange acellular material (a.k.a. concretions), (Figure 4). Other areas of the liver lack fibrosis but show prominent steatotic change in periportal distribution (Figure 5).
|
|
Figure 3. Low power view shows irregular islets of hepatocytes encircled by dense fibrosis and numerous proliferating ductules (orange arrows). |
|
|
Figure 4. Higher magnification shows ductules expanded by pink and light-orange concretions (orange arrows). |
|
|
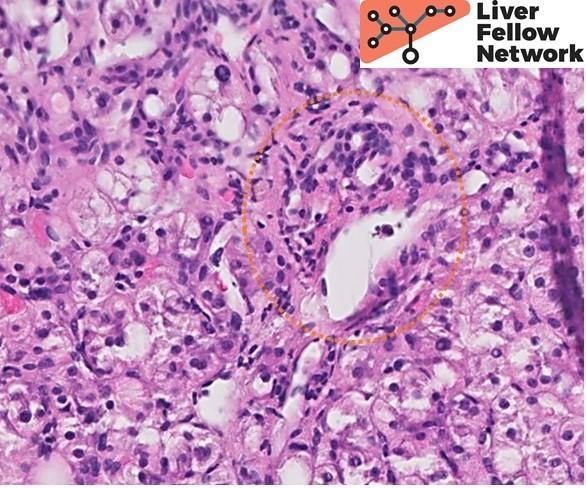
Figure 5. Non-nodular parenchyma shows numerous steatotic hepatocytes encircling the portal tract (orange circle). |
In light of the clinical history and the presence of bi-allelic CFTR mutations, the findings are consistent with hepatic involvement by cystic fibrosis.
Cystic Fibrosis
Definitions and pathogenesis
Cystic Fibrosis (CF) is an autosomal recessive disorder of dysfunctional chloride transport affecting the biliary tree, the bronchial, salivary and intestinal glands, and the exocrine pancreas. It is estimated to affect 1 in 2,500 liver births in White populations worldwide. It is caused by a large spectrum of different alterations in CFTR gene encoding cystic fibrosis transmembrane regulator, a cAMP-dependent chloride channel located in the apical membrane of secretory epithelial cells in various tissues, including branching hepatic bile ducts and the gallbladder. The pathogenic CFTR alterations are subdivided into 5 functional classes, with the most common being a class II alteration, F508del, which leads to accumulation of CFTR protein in the endoplasmic reticulum. Hepatobiliary involvement appears to be associated with class I-III mutations that impair synthesis, intracellular trafficking or regulation of CFTR. The main functions of this protein are to mediate transmembrane efflux of chloride ions and to regulate other ion channels and cellular processes. Consequently, dysfunction of CFTR leads to physicochemical changes in secretions within the tissues involved, complicated by ductal obstruction and secondary gland dysfunction. Specifically in the liver, CFTR protein is expressed on the apical membrane of cholangiocytes and regulates the content of water and electrolytes of bile. Loss of function results in production of thick, viscous bile with decreased alkalinity leading to impaired secretion, increased susceptibility to infections, and injury to the surrounding tissue from a combination of factors including direct bile-induced toxicity, associated release of free radicals, and secondary inflammation and fibrosis.
Clinical characteristics
Clinically significant liver involvement occurs in approximately 4-6% of CF patients, although up to half of patients can show asymptomatic biochemical abnormalities. The clinical presentation of hepatobiliary disease is variable. Infants may present with neonatal conjugated hyperbilirubinemia, often in context of a meconium ileus, or symptoms of bile duct obstruction. In older children, asymptomatic hepatosplenomegaly with intermittent transiminitis and elevated ALP may be the clinical presentation. A minority of patients demonstrate a progressive disease leading to hepatic cirrhosis and complications such as portal hypertension, hypersplenism and variceal bleeding. Cholelithiasis is another potential hepatobiliary manifestation.
Diagnostic Modalities
The diagnosis of CF requires presence of characteristic symptoms involving at least one organ system, positive newborn screen or sibling with CF and demonstration of CFTR dysfunction. Newborn screening is performed routinely in North America and comprises a combination of genetic methods and immunoassays. If positive, patients should undergo a sweat chloride test which can identify increased chloride concentration seen in CF patients. Sweat chloride ≥60 mmol/L is diagnostic of CF if confirmed by a repeat test or DNA analysis. The advantage of CFTR sequencing as a diagnostic modality is its ability to both confirm the diagnosis and genotype the patient which is valuable for therapeutic guidance and prognostication. Other modalities include nasal potential difference measurements and organ system-specific assessments.
Pathologic Features
Liver biopsies from neonates will most commonly show a nonspecific neonatal cholestasis pattern secondary to mucus impaction of extrahepatic and intrahepatic bile ducts. The findings include variably prominent cholestasis accompanied by lobular and portal inflammation, giant cell transformation, hepatocyte ballooning and acidophil bodies (Figure 6). Extramedullary hematopoiesis is commonly seen. Neonatal cholestasis has a wide differential, including biliary atresia, infections, drugs and total parenteral nutrition, and a wide array of metabolic disorders such as alpha-1-antitrypsin deficiency, glycogen storage disorders, and progressive familial intrahepatic cholestasis. Consequently, these histologic findings need to be contextualized with respect to the clinical, radiologic, laboratory and genetic findings.
|
|
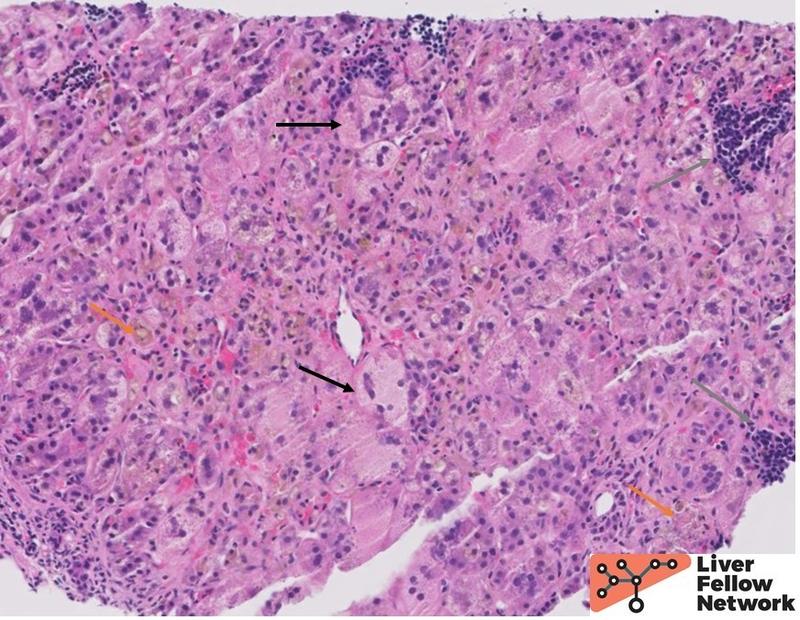
Figure 6. A biopsy from a patient with CF shows inflamed liver parenchyma with prominent hepatocellular and canalicular cholestasis (orange arrow), giant cell transformation and ballooning (black arrow), and patchy extramedullary hematopoiesis (gray arrow). |
Hepatic steatosis is the most common pathologic finding in older children and adults, likely resulting from a combination of malnutrition, abnormal fatty acid metabolism, oxidative stress and insulin resistance. Unlike alcohol-related and unrelated fatty liver disease, steatosis is more commonly periportal, rather than centrilobular. Periportal hemosiderin can also be found, likely due to increased iron transport in context of pancreatic insufficiency.
Up to a third of CF patients will have focal biliary cirrhosis. This pattern is characterized by irregular portal and septal fibrotic scars accompanied by prominent ductular reaction. As seen in this case, the ductules are characteristically filled with pink to light-orange amorphous concretions, pathognomonic of CF. Some ductules can rupture and induce an inflammatory response and occasionally be associated with a superimposed bacterial infection. Progression of fibrosis will eventually lead to multilobar biliary cirrhosis.
Gallbladder findings include cholelithiasis, cholecystitis and gallbladder mucous cysts.
Treatment and prognosis
Management of CF hepatobiliary disease requires a combination of risk reducing and therapeutic approaches. Nutrition optimization and supplementation are critical to counterbalance the bile acid abnormalities in CF patients. Risk factors for other hepatobiliary diseases, such as alcohol or viral hepatitis, should be minimized as well. Use of ursodeoxycholic acid remains controversial, although some clinical evidence suggests an associated improvement of biochemical parameters. Approximately 5% of CF patients will eventually progress to cirrhosis which can be complicated by portal hypertension. In these instances, portosystemic shunt and liver transplantation are therapeutic considerations.
References
- Burt, MacSween's Pathology of the Liver: Eighth Edition. 2022.
- Tsui LC. The spectrum of cystic fibrosis mutations. Trends Genet 1992;8:392-8.
- Duthie A, Doherty DG, Williams C, et al. Genotype analysis for delta F508, G551D and R553X mutations in children and young adults with cystic fibrosis with and without chronic liver disease. Hepatology 1992;15:660-4.
- McKone EF, Emerson SS, Edwards KL, et al. Effect of genotype on phenotype and mortality in cystic fibrosis: a retrospective cohort study. Lancet 2003;361:1671–6.
- Herrmann U, Dockter G, Lammert F. Cystic fibrosis-associated liver disease. Best Pract Res Clin Gastroenterol 2010;24:585–92.
- Kobelska-Dubiel N, Klincewicz B, Cichy W. Liver disease in cystic fibrosis. Prz Gastroenterol 2014;9:136-141.