Hepatobiliary Disease in Cystic Fibrosis: Where Are We Now?

Cystic fibrosis is an autosomal recessive disease secondary to a mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Typical manifestations of CF include chronic lung infections, pancreatic insufficiency, sinusitis, and infertility. CF is often associated with shortened lifespans, and the most common cause of death is end-stage lung disease. However, with advancements in treatment like CFTR modulators, people with CF (pwCF) are living longer and other disease manifestations will likely continue to increase in importance including a variety of hepatobiliary pathologies.
The hepatobiliary pathologies seen in CF range from cholelithiasis, hepatic steatosis, liver fibrosis to multilobular cirrhosis, portal hypertension, liver failure necessitating liver transplant, or even liver-related death. Recent guidelines (2023) for screening, evaluation, and management of hepatobiliary disease for pwCF has been published to improve early diagnosis and care for CF-associated hepatobiliary disease and ultimately improve morbidity and mortality.
Throughout the years, there have been multiple published definitions for CF liver disease (see table below). In 2024, ESPGHAN and NASPGHAN published a joint position paper on standardizing the classification of CF liver disease.

Terminology for CF- associated hepatobiliary disease in 2024
- Advanced CF liver disease (aCFLD) refers to having one (or more) of the following: nodular liver, advance fibrosis (F4), multilobular cirrhosis with or without portal hypertension, or noncirrhotic portal hypertension.
- CF hepatobiliary involvement (CFHBI) refers to having one (or more) of the following without features of aCFLD: hepatomegaly, liver fibrosis (<F4), increased liver stiffness by elastography (<F4), hepatic steatosis, focal biliary cirrhosis, cholestasis, persistent (>3-6 months) elevated serum liver function tests (any level above upper limit of normal), abnormal liver imaging, cholelithiasis, sclerosing cholangitis, or hepatolithiasis.
Biochemical Findings of Hepatobiliary Involvement in pwCF
Elevation of Liver Enzymes
There is an ongoing debate if elevated liver enzyme levels are useful biomarkers/predictors of liver involvement in pwCF. Thus, the findings of elevated liver enzymes in pwCF should be evaluated in consideration of other factors like recurrent respiratory infections, nutritional issues, and potential drug-induced liver injury (ex. CFTR modulators). Persistent elevation of transaminases is a common manifestation of CFHBI and should warrant a broader diagnostic approach to the underlying cause.
Although elevated liver enzymes in pwCF are neither sensitive nor specific in the detection of CFHBI, there is value in utilizing combination measurements that incorporate liver enzymes with other measurements (e.g. APRI, FIB-4, and GPR) in detecting aCFLD and cirrhosis.
The CF Foundation currently recommends annual calculation of at least one liver fibrosis index in pwCF to monitor liver disease progression. Biopsy-validated studies in pediatric CF liver disease show that APRI is superior to FIB-4 in differentiating CF liver disease from CF without liver disease and for predicting severe fibrosis. APRI scores above a cutoff of 0.462 were a significant indicator of severe fibrosis. However, these values should be interpreted in the context of clinical findings, imaging, and serial trends over time to guide more advanced testing and management.
Gamma-glutamyl transpeptidase (GGT)
GGT is expressed in the cholangiocytes and an increase in serum GGT may indicate bile duct or cholangiocyte damage. In children with CF, a persistent elevation of GGT is defined as a value above 30 IU/L. A sustained elevation of GGT > 30 IU/L has been associated with the presence of structural nodular liver transformation and cirrhosis.
Hepatic Imaging
Liver ultrasound (US) is currently the most frequently used and advised imaging method for characterizing the liver in pwCF. The use of US has not been recommended universally for routine screening but has been recommended if there is a suspicion for liver involvement based on physical exam and laboratory findings. Sellers et al (2019) demonstrated that targeted use of US based on indices of fibrosis can increase the detection of advanced liver disease in children with CF. In addition, the article recommends that pwCF and CFHBI should have an abdominal US performed at least every 2 years to monitor disease progression.
Patients with heterogenous pattern of the liver on US had a 5.2-fold increased incidence of cirrhosis and a 6.1-fold increased incidence of portal hypertension compared to children with normal pattern on liver US. The findings of nodular liver on US have a good specificity for structural liver disease in adults with CF. However, it is difficult to distinguish between cirrhotic or non-cirrhotic disease of the liver in those with this nodular pattern.
Hepatic Histopathology
While liver biopsy is considered the gold standard for diagnosing and categorizing liver disease, it is not commonly used in CF care. The refrain from liver biopsy is secondary to the limited clinical and therapeutic consequences of the histological results and potential increased procedural risks due to pulmonary hyperinflation. Also, histological abnormalities can be focally distributed in the liver and introduce sampling error. Common indications for liver biopsy in pwCF include:
- Need for differentiation between cirrhotic and non-cirrhotic liver disease
- Evaluation of other causes of liver disease that are potentially treatable
- Evaluation of liver-lung transplantation candidates regarding clinical consequences and therapeutic options
CF has been associated with 4 primary hepatic histopathologic patterns:
- Preponderant fibrotic and cirrhotic features
- Multilobular cirrhosis
- Focal biliary cirrhosis
- Portal or cholangial inflammation with fibrosis
- Obliterative portal venopathy
- Steatosis with or without inflammation or fibrosis
- (Neonatal) cholestatic features
Liver Stiffness Measurements (LSM)
LSM is used to determine the elastic properties of the liver. An increase in liver stiffness correlates with fibrotic changes of the liver parenchyma. There are multiple modalities that can be used to measure liver stiffness, this includes transient elastography, 2D shear wave elastography, US elastography, and magnetic resonance elastography (MRE). Due to the variation of technology, cutoff values for LSM are modality specific.
Due to the inconsistent CF liver disease definitions over time, there is a wide range of collective experience with liver stiffness measurements and thus cutoff values.
Overall, LSM can be used as a quantitative measurement to grade the degree of liver fibrosis in pwCF and help monitor for the progression of liver disease.
Presence of Portal Hypertension (PHT)
Liver involvement presenting with PHT is the most clinically significant manifestation of CFHBI associated with increased morbidity and mortality. The most common sign of PHT in pwCF is splenomegaly, which is identified by physical exam or imaging. The use of imaging to assess for splenomegaly is preferred because of thoracic hyperinflation, which may result in a more readily palpable spleen. Thrombocytopenia can also be seen and is strongly associated with PHT. It is advised to monitor platelets overtime as their levels can be impacted by inflammatory processes and infections. Gastrointestinal variceal bleeding can be a more advanced sign of PHT and may be confirmed through endoscopic diagnosis.
CFHBI Classification based on biochemistry, imaging, histology, LSM, and clinical signs as proposed by ESPGHAN/NASPHGHAN in 2024.

Treatment
There have been several small randomized controlled trials and multiple cohort studies in pwCF liver disease that have shown improvements in liver enzymes (AST, ALT, and GGT) with ursodeoxycholic acid (UDCA) treatment. However, no studies have shown that UDCA prevents the development of aCFLD or progression to liver transplant. In 2017, Cochrane Review concluded that there was insufficient evidence to recommend routine UDCA use in CF. Thus, the CF foundation recommends against the routine use of UDCA.
The use of CFTR modulator treatment should be closely monitored by hepatologists because the benefits to CF lung disease outweigh the liver-related risk. CFTR modulators can be liver toxic and 5% of patients develop liver enzyme elevations (> 3x ULN), although very few require treatment discontinuation. Treatment interruption and rechallenge is sufficient in most cases.
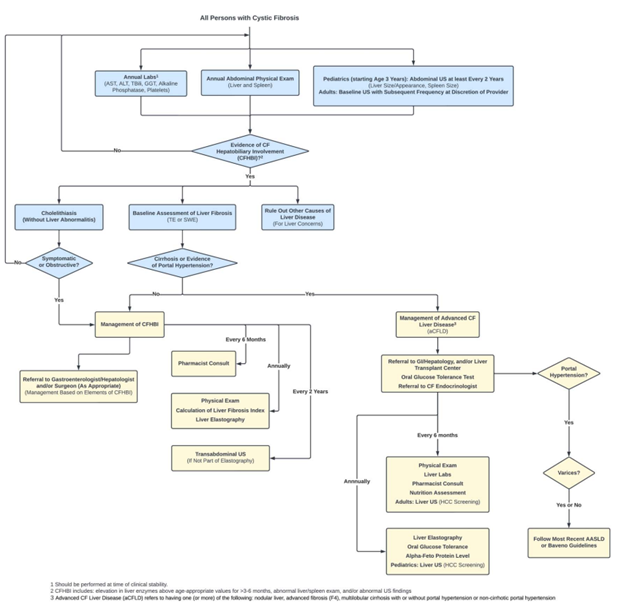
Monitoring and Screening Recommendations by the CF Foundation
Overall, there are still many research gaps that remain in CF liver disease. As new data emerges, and the use of CFTR modulators increases these recommendations may need to be revisited. These current recommendations provide a good roadmap to earlier detection and monitoring of CFHBI and aCFLD.