Level Up on Ammonia and Encephalopathy

Learning Objectives
By the end of this article, readers should be able to:
- Describe the physiology, metabolism, and excretion of ammonia.
- Differentiate between hepatic and non-hepatic causes of hyperammonemia.
- Explain the pathophysiology of hyperammonemia-induced encephalopathy and outline key management strategies.
Introduction
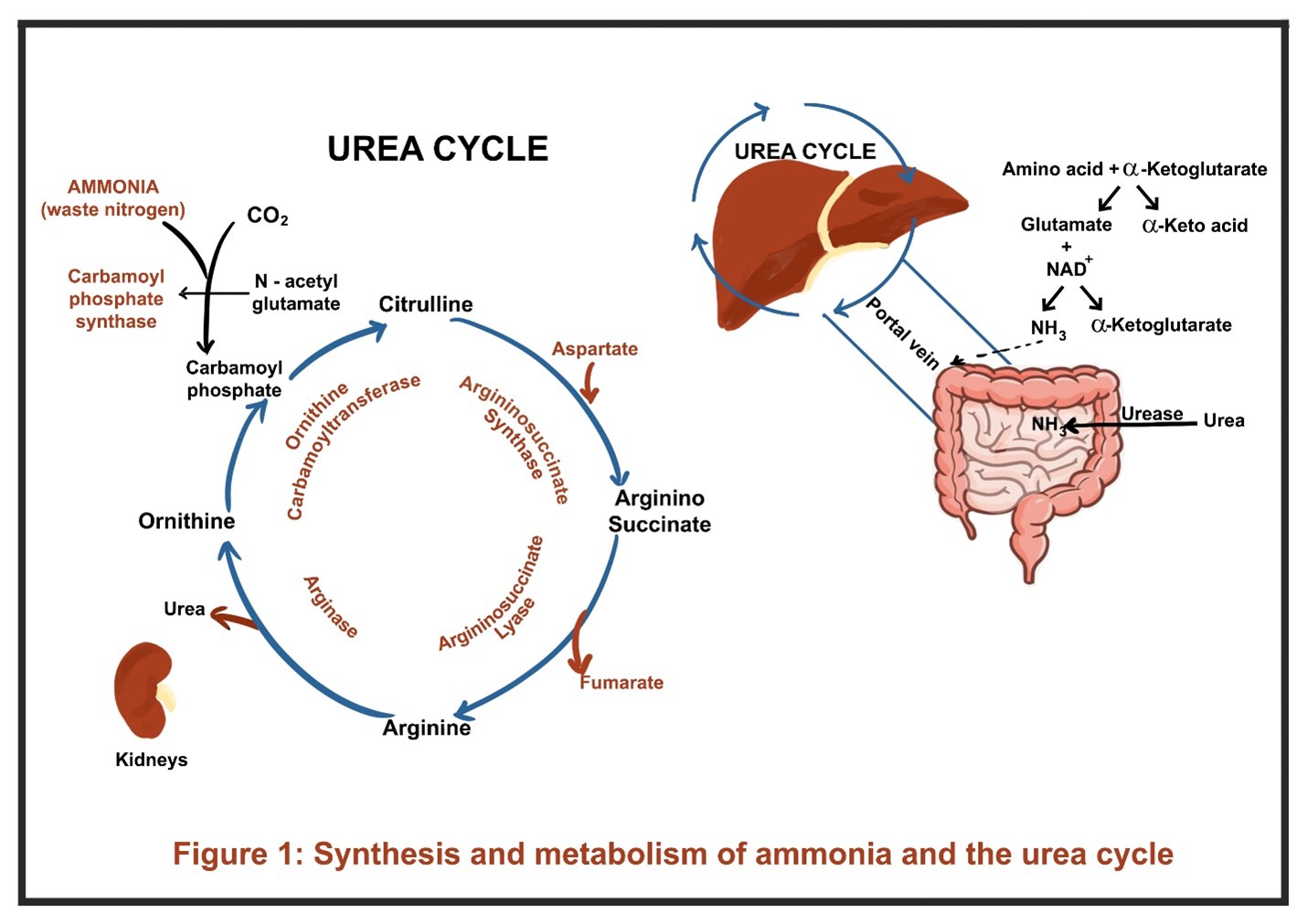
Ammonia is a small, nitrogen-containing molecule (NH₃) that is produced in the body primarily as a byproduct of protein metabolism. It cannot be directly excreted in the urine and is metabolized to water-soluble urea via the urea cycle in the liver, which is then renally eliminated. When these pathways are impaired, ammonia accumulates in the blood, leading to hyperammonemia.
Ammonia Synthesis, the Urea Cycle, and Excretion
Ammonia is produced by several metabolic processes:
- Skeletal muscle generates ammonia during amino acid breakdown.
- Intestinal bacteria degrade proteins and urea through urease activity, releasing ammonia.
- Kidneys generate ammonia from glutamine through renal ammoniagenesis. In the proximal tubules, ammonia combines with hydrogen ions to form ammonium (NH4+), which is then excreted in urine.
Under normal physiological conditions, ammonia is rapidly detoxified by two main pathways: 1) the urea cycle, and the 2) glutamine synthetase reaction.
- Ammonia travels via the portal circulation to the liver, where the urea cycle converts it into urea, a water-soluble and less toxic molecule (Figure 1). Urea is then transported to the kidneys and excreted.
- At a biochemical level, amino acids undergo transamination with alpha-ketoglutarate to form glutamate. Glutamate is then deaminated by glutamate dehydrogenase, releasing ammonia for entry into the urea cycle.
- Extrahepatic detoxification occurs in tissues such as muscle and brain. Ammonia is bound to glutamate by glutamine synthetase to form glutamine. Glutamine is later transported to the kidneys, where it undergoes ammoniagenesis, eventually producing ammonium for urinary excretion.
What is the role of ammonia?
Ammonia is essential for several physiological functions:
- Key nitrogen donor for synthesis of amino acids and nucleotides.
- Acid-base balance, particularly through renal ammoniagenesis and urinary ammonium excretion. In metabolic acidosis, the kidneys generate ammonia from glutamine metabolism in the proximal tubule, facilitating net acid excretion while generating bicarbonate.
- Neurotransmitter metabolism, particularly in the regulation of glutamate and GABA via the glutamine synthetase pathways in astrocytes.
Prevalence of Hyperammonemia
In neonates, inherited metabolic diseases such as urea cycle disorders (UCDs) and organic acidemias are the most common causes. Inherited UCDs occur in about 1 in 35,000 births. In adults, hyperammonemia is most often related to liver disease. Hepatic encephalopathy (HE), affects about 30% of patients with cirrhosis. In critically ill adults admitted to the ICU, hyperammonemia (not due to liver failure) was observed in approximately 4.5% of patients in a large case series.
2. What are the hepatic and non-hepatic causes of hyperammonemia?

A. Congenital Causes
UCDs are inborn errors of nitrogen detoxification caused by defects in the urea cycle enzymes or transporters. They typically present in the neonatal period, most often within the first few days after birth in cases of severe enzyme deficiency. These neonates appear normal at birth but rapidly develop signs of hyperammonemia such as poor feeding, vomiting, lethargy, and progression to coma if untreated.
In some cases, the first signs of UCDs, including episodes of encephalopathy, may not appear until adulthood and are usually milder. For this reason, clinicians should remain alert to possible symptoms in adults to ensure timely diagnosis. Adult presentations are nonspecific and may include headaches, avoidance of protein-rich foods, mood or psychiatric changes after intense exercise or childbirth, attention difficulties, fatigue, and seizures. Stressors such as surgery, bleeding, total parenteral nutrition, and starvation can precipitate hyperammonemia in patients with partial enzyme activity. Some UCDs can be confirmed by measuring metabolites, whereas others require genetic testing. The key laboratory finding is high serum ammonia levels. Hyperammonemia may be defined as a plasma ammonia level above 50 μmol/L in term infants, children, and adults, and above 100 μmol/L in neonates. The linked review article describes presentation and management of adults with UCD.
Common UCDs include are included in Table 1.
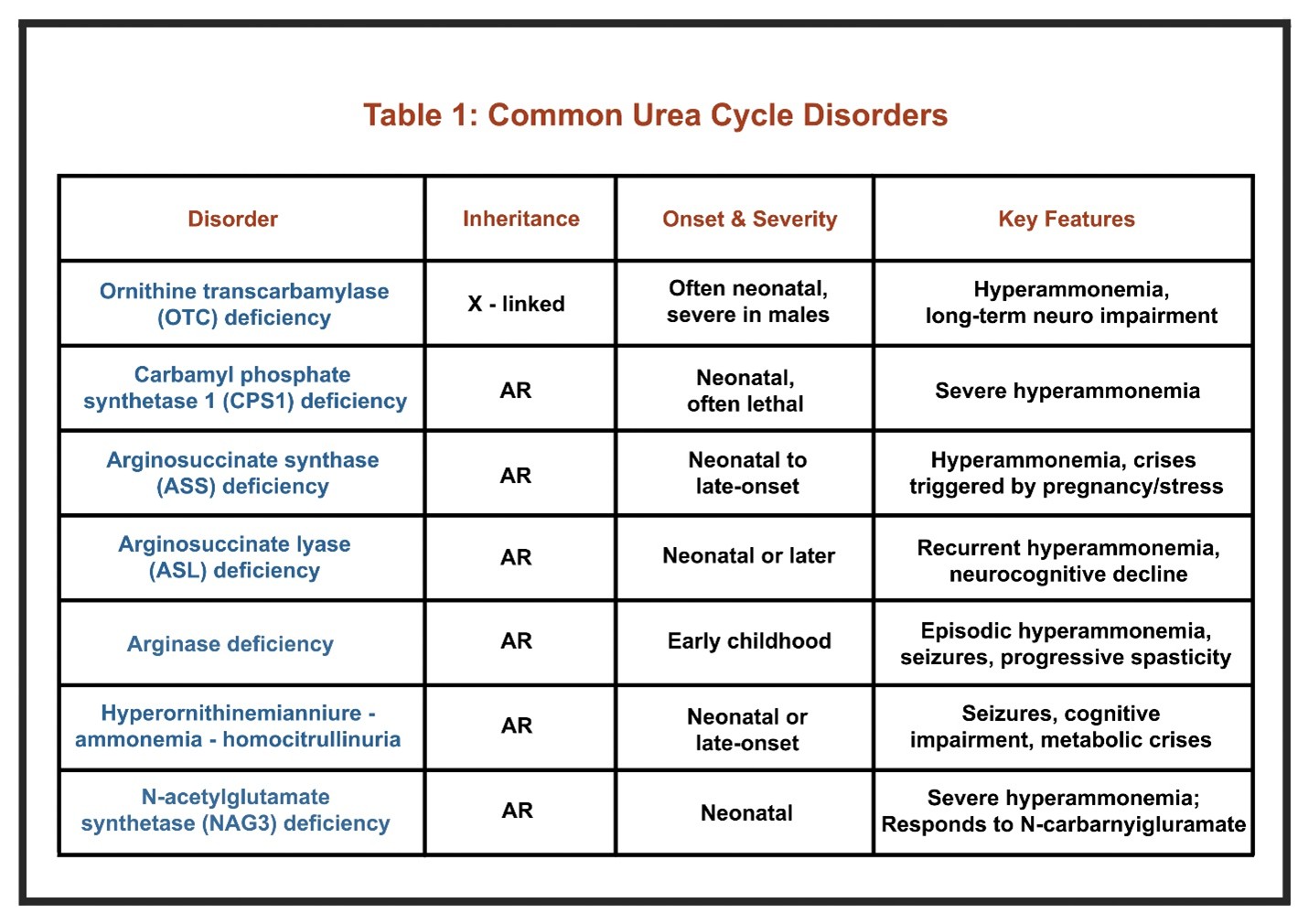
More information about UCDs can be found here.
Organic Acidemias and fatty acid oxidation defects:
In organic acidemias, the urea cycle is inhibited through the buildup of organic acid metabolites, thus impairing ammonia detoxification.
In fatty acid oxidation disorders, impaired mitochondrial β-oxidation leads to reduce ATP availability. The energy deficit compromises the function of urea cycle enzymes, leading to subsequent buildup of ammonia.
B. Acquired causes of Hyperammonemia
Non-Hepatic
- Hematological malignancies: Hyperammonemia is a rare but serious complication with hematological malignancies, most commonly occurring after intensive chemotherapy, hematopoietic stem cell transplantation, or asparaginase therapy.
- Medications:Valproate inhibits CPS1; 5-fluorouracil reduces ATP through Krebs cycle inhibition. Other agents, including gemcitabine, cisplatin, and tyrosine kinase inhibitors, having unclear mechanisms.
- Urinary tract infections: Urease-producing organisms break down urea into ammonia, which can diffuse into the blood stream. These include Escherichia coli, Klebsiella species, Proteus mirabilis, Morganella morganii, Providencia rettgeri, diphtheroids, and Herpes simplex.
- Gastrointestinal causes: Digestion of blood in the GI tract delivers a large protein load, primarily hemoglobin, to the gut. Hemoglobin is catabolized by intestinal mucosal enzymes and colonic bacteria, generating substantial amounts of ammonia as a metabolic byproduct.
- Bariatric surgery may unmask UCDs, induce catabolism, or alter the microbiome, all of which contribute to hyperammonemia. After procedures such as gastric bypass, altered gastrointestinal anatomy can result in inadequate protein intake and absorption, leading to reduced hepatic synthesis of urea cycle enzymes and cofactors, impairing ammonia detoxification. Micronutrient deficiencies, especially zinc, contribute to decreased activity of urea cycle enzymes.
- Urinary diversions: Procedures such as ureterosigmoidostomy, ileocecal pouch, and ileal conduit can promote ammonia absorption in the presence of urease-producing bacteria.
- Renal disorders: Distal renal tubular acidosis (Type I), Type IV RTA, Chronic kidney disease and renal failure, impair renal ammoniagenesis and excretion.
Hepatic Causes of Hyperammonemia
Acute liver failure: In acute liver failure, hepatocellular dysfunction and loss of functional hepatic mass result in a marked reduction in urea cycle activity, causing ammonia to accumulate in the systemic circulation. The consequences of rapid rise in ammonia levels are described below.
Cirrhosis: As fibrosis and nodular regeneration distort hepatic architecture in cirrhosis, portal hypertension develops, resulting in the formation of portosystemic collateral vessels. These shunts allow ammonia-rich portal blood to bypass the liver and enter the systemic circulation directly, further elevating systemic ammonia levels.
3. Why does elevated ammonia cause neurological dysfunction?
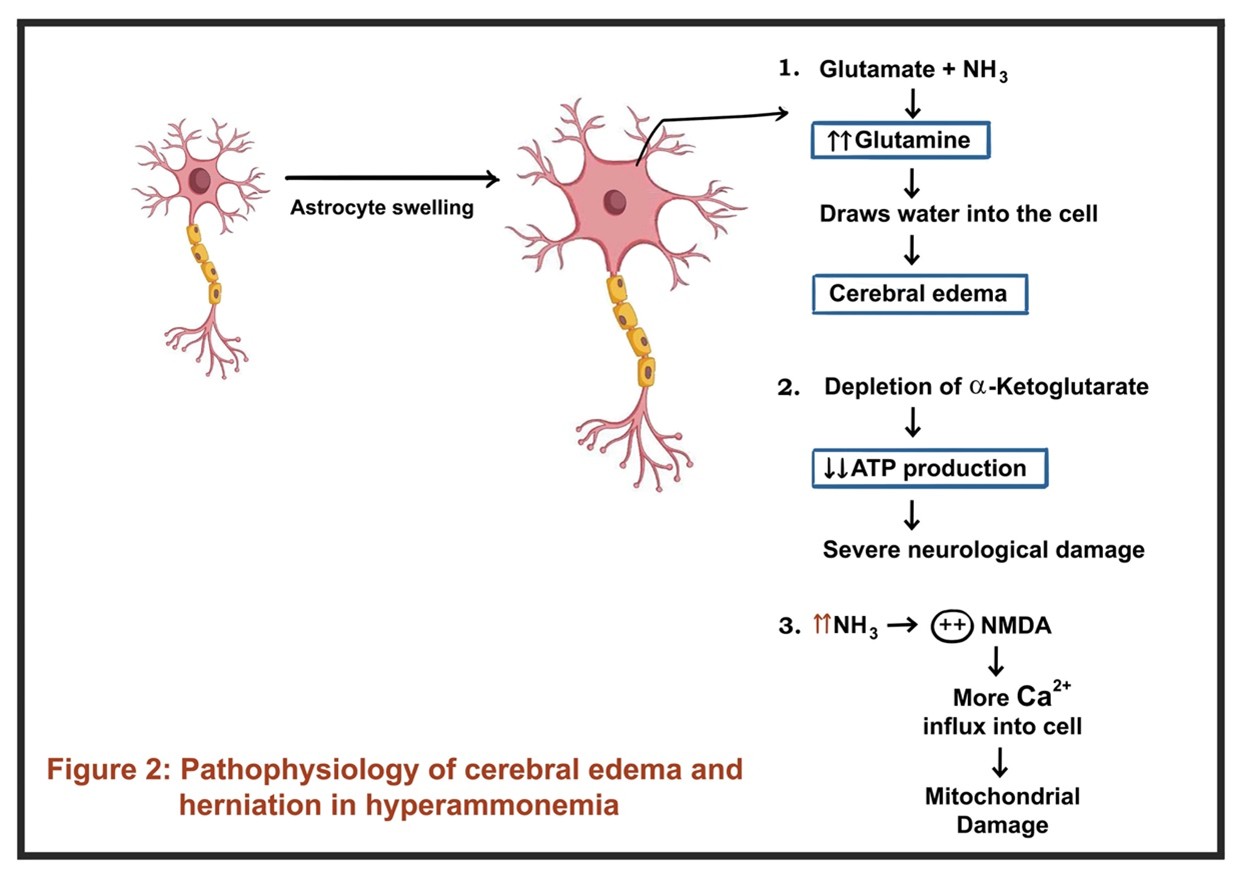
In acute liver failure (ALF), the rapid loss of the hepatic detoxification capacity leads to a sudden and marked rise in systemic ammonia. Ammonia crosses the blood-brain barrier and is metabolized by astrocytes to glutamine. This glutamine acts as an osmolyte, causing astrocytic swelling and cytotoxic cerebral edema, which can rapidly progress to herniation. Hyperammonemia (>150 μmol/L) is a risk factor for intracranial hypertension. Given the acute rise in ammonia, compensatory mechanisms (e.g., upregulation of ammonia transporters, osmolyte regulation) are not established in ALF, making the brain highly susceptible to ammonia-induced injury and edema.
In chronic liver failure (cirrhosis), ammonia levels are often elevated but typically rise more gradually. The brain adapts over time through osmotic and neurotransmitter changes, reducing the risk of acute cerebral edema. Neurological dysfunction manifests as HE. Chronic exposure leads to astrocytic changes and altered neurotransmission. Ammonia acting synergistically with other neurotoxins and inflammatory mediators. GABAergic tone is increased and glutamatergic signaling is altered, leading to global slowing of brain activity and cognitive, motor, and perceptual disturbances.
4. Clinical Presentation and Management
The clinical presentation of hyperammonemia is characterized by progressive neurological dysfunction. Early symptoms include subtle behavioral changes, anorexia, irritability, vomiting, lethargy, and somnolence. As ammonia levels rise, patients may develop disorientation, negative myoclonus (manifesting as asterixis and/or milk maids grip), ataxia, tremors, seizures, and abnormal respiratory patterns (hyperventilation or hypoventilation). In severe cases, hyperammonemia leads to coma, cerebral edema, and death.
Diagnostic Considerations
- Ammonia levels do not consistently correlate with the severity of HE. Rather, HE is a clinical diagnosis. Sample handling, fasting state, and processing delays can significantly affect accuracy. Check out this LFN Why Series Article for more on why ammonia should not be used in the diagnosis and management of HE.
- Altered mental status in cirrhosis cannot always be attributed to hepatic encephalopathy. Check out this LFN Back to Basics Article, for looking at common triggers and mimickers for HE.
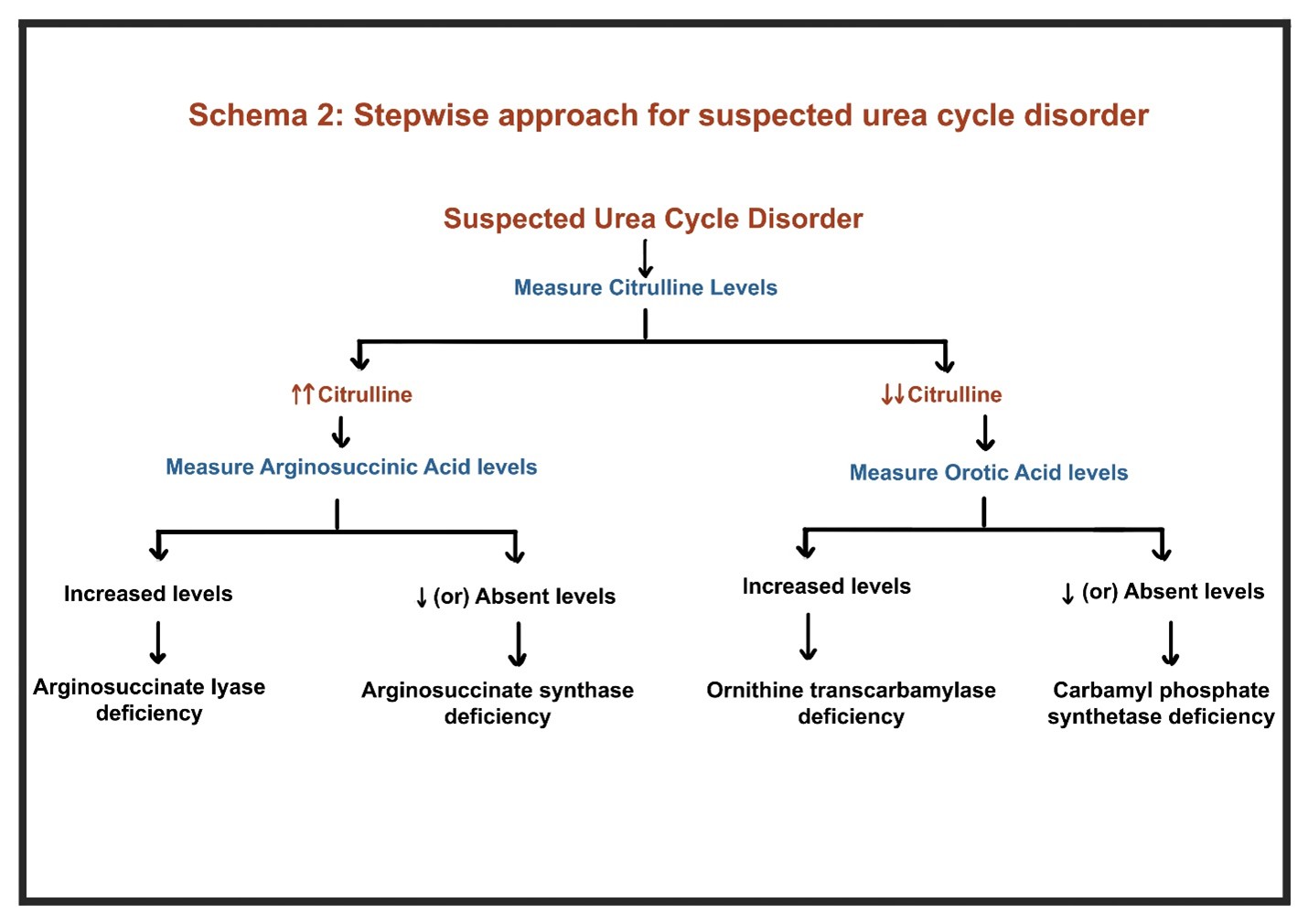
- In patients without liver disease, stepwise evaluation should include arterial pH, serum ketones, glucose, amino acid profiles, and genetic testing for UCDs (Schema 2)
Management Principles
Management of HE has been covered in depth in previous LFN posts, including in the Back to Basics Series, and Why Series. Basic principles include stopping offending drugs, treat infections/precipitants, and initiate lactulose ± rifaximin as first-line; emerging options include L-ornithine L-aspartate (LOLA), nitrogen scavengers. Imaging for large portosystemic shunts and IR closure should be considered in unexplained or refractory HE. Renal replacement therapy is used for severe/refractory hyperammonemia.
Summary
Ammonia is a byproduct of protein metabolism, detoxified mainly by the liver through the urea cycle and glutamine synthesis. Hyperammonemia occurs when these pathways are impaired, due to congenital or acquired hepatic and non-hepatic causes. It is important to get a detailed history, and consider the various causes of hyperammonemia, especially in a patient presenting for the first time, without obvious liver disease. Elevated ammonia disrupts brain function via astrocytic glutamine accumulation, neurotransmitter imbalance, and cerebral edema. Management focuses on correcting triggers, reducing ammonia production, enhancing excretion, and in severe cases, using dialysis or shunt embolization
Take-Home Points
- Ammonia physiology: Generated in muscle, gut, and kidneys, then detoxified by the urea cycle and glutamine synthetase pathways.
- Etiologies: Can be hepatic (cirrhosis, acute liver failure) or non-hepatic (urea cycle disorders, drugs, infections, metabolic conditions).
- Pathophysiology: Acute increases cause cerebral edema (especially in ALF), while chronic exposure alters neurotransmission and cognition (HE in cirrhosis).
- Clinical features: Range from subtle cognitive changes to seizures, coma, and intracranial hypertension.
- Management: Identify and treat precipitants, use lactulose and rifaximin as first-line, and renal replacement therapy in severe cases.
- Pearl: Ammonia levels do not reliably correlate with encephalopathy severity. Assess clinically, correct precipitating factors and exclude mimickers.