The Real Deal Behind Alagille Syndrome

The Real Deal Behind Alagille Syndrome
Background
Alagille syndrome is an autosomal dominant, multisystem disorder with characteristic hepatic, cardiac, vascular, renal, skeletal, ocular, and facial manifestations. It was first described by French hepatologist Daniel Alagille in 1969 in a case series describing 30 patients with hypoplastic intrahepatic bile ducts with characteristic extra-hepatic clinical features.1The incidence of Alagille syndrome is currently thought to be approximately 1:30,000 live births.
Genetics
Pathogenic mutations in the JAGGED1 (JAG1) gene, encoding a ligand in the Notch signaling pathway have been identified in approximately 89% of Alagille syndrome cases. A smaller subset (2.5%) have a pathogenic variant in NOTCH2, which encodes the Notch signaling receptor. Both genes disrupt Notch signaling and influence downstream gene expression. In a small percentage of patients (~3%), genetic testing does not reveal a molecular diagnosis despite the patient meeting criteria for a clinical diagnosis.
Clinical Features
There are no observed genotype-phenotype correlations in Alagille syndrome and multiple case reports have described significant variation in the clinical presentations within families with the same pathogenic variant. Patients with severe liver or cardiac manifestations are often diagnosed with ALGS in infancy, whereas those with more subtle manifestations may not have an established diagnosis until much later in life (sometimes adulthood).
Figure 1: Clinical Features of Alagille Syndrome
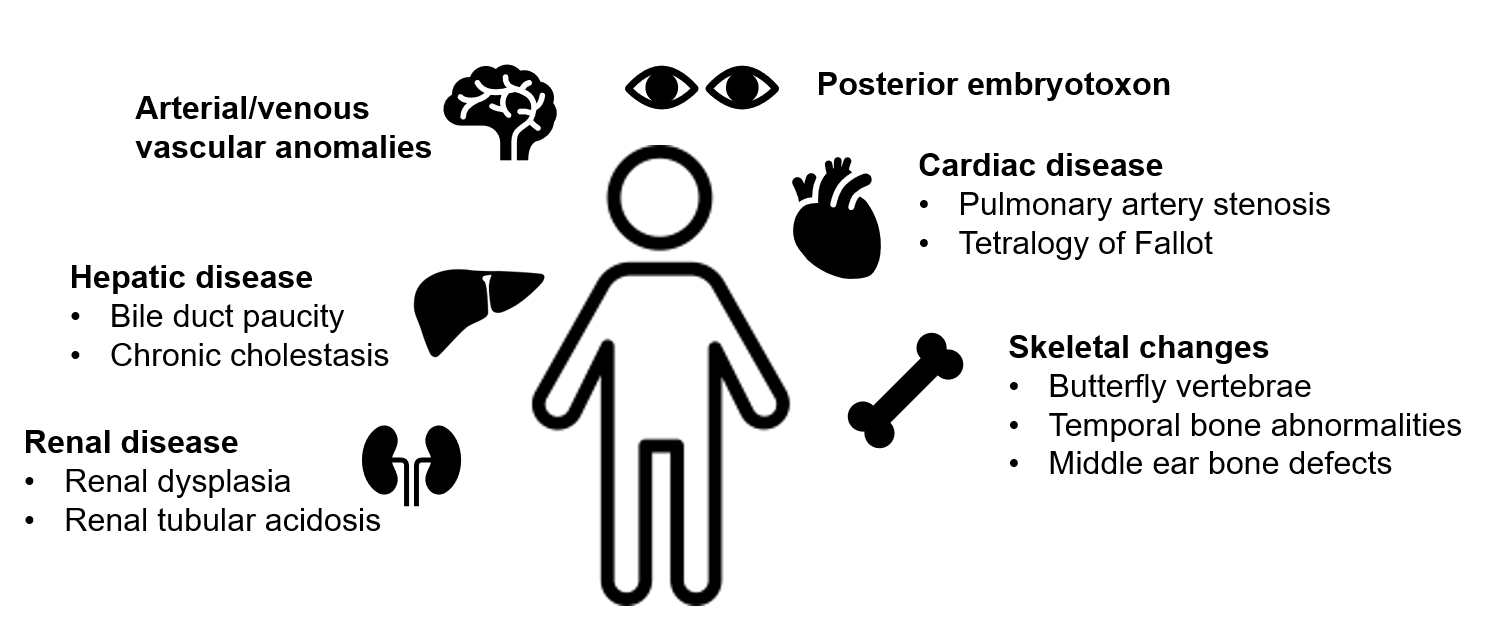
Hepatic
Liver disease in Alagille syndrome is characterized by cholestasis, with bile duct paucity on liver biopsy. Bile duct paucity is defined as a decreased bile duct-to-portal tract ratio (< 0.5, compared to normal ratio of 0.9-1.8). Given post-natal development of bile ducts and subsequent progression of duct paucity over the first months of life, findings may not be detectable on liver biopsy prior to age 6 months. In children under 5 years of age, total bilirubin > 6.5 mg/dL, conjugated bilirubin > 4.5 mg/dL, and cholesterol > 520 mg/dL have been identified as predictors of sustained and more severe liver disease.
Cholestasis typically manifests as unremitting pruritus, which can develop early and become one of the most debilitating symptoms of Alagille syndrome. Xanthomas can also develop due to decreased cholesterol secretion and can be disfiguring.
Benign and malignant hepatic lesions are a rare complication of Alagille syndrome. Benign, homogenous regenerative nodules with relative preservation of interlobular bile ducts do not require intervention. Hepatocellular carcinoma (HCC) can rarely present as single or multifocal heterogenous tumors prone to necrosis, hemorrhage, and metastasis. The risk for developing HCC regardless of disease severity highlights the need for a cancer screening protocol to facilitate early detection and treatment.
Cardiac
Cardiac abnormalities can be seen in up to 94% of patients with Alagille syndrome, most commonly branch pulmonary artery stenosis and tetralogy of Fallot (TOF). Survival in Alagille syndrome patients is impacted by the presence of significant intracardiac heart disease (such as TOF, critical pulmonary atresia), with 6-year survival decreased to 40% compared with 95% in patients without intracardiac disease.
Vascular
Arterial and venous vascular anomalies have been reported in Alagille syndrome. These can be detected within the first decade of life and are a significant cause of morbidity and mortality. Intracranial bleeding is the most common vascular complication, occurring in 14 percent of patients. A screening brain MRI/MRA is recommended for all Alagille syndrome patients once they are developmentally appropriate to obtain imaging without the need for sedation.
Renal
Renal involvement, including renal dysplasia and renal tubular acidosis, has been described in 39% of patients with Alagille syndrome. Patients should have a complete renal function and structure evaluation at the time of diagnosis, and with liver transplantation evaluation if needed.
Skeletal
Butterfly vertebrae are the most common skeletal abnormality. This finding is not of structural significance but can help with making the diagnosis of Alagille syndrome. Other skeletal abnormalities include temporal bone abnormalities and middle ear bone defects, which can lead to chronic otitis media and hearing loss.
Facial
The characteristic facial features associated with Alagille syndrome include triangular facies with prominent forehead, deeply set eyes, moderate hypertelorism, bulbous nose, and a pointed chin. Facial features have been identified as the clinical feature with highest penetrance, with almost universal occurrence in JAG1 mutation-positive probands and relatives.
Ocular
Posterior embryotoxon refers to a prominent, centrally positioned ring or line at the Schwalbe ring (the point where the corneal endothelium and uveal trabecular meshwork join) and is the most common ocular feature in Alagille syndrome. Other findings such as iris abnormalities, optic disc or fundus changes have also been reported. Like the skeletal findings, posterior embryotoxon is of limited clinical significance and does not typically affect vision but can assist with diagnosis.
Growth Failure
Poor growth and neurocognitive deficits are widely seen in Alagille syndrome. Growth failure is likely multifactorial in etiology, thought to be secondary to increased caloric demands, fat malabsorption from cholestasis, and the potential role of JAG1 in growth deficiency. Approximately half of Alagille syndrome patients require special education services for neurocognitive delay. Improvements in nutritional status and underlying cholestasis may help combat neurocognitive impairment.
Table 1: Clinical Features of Alagille Syndrome
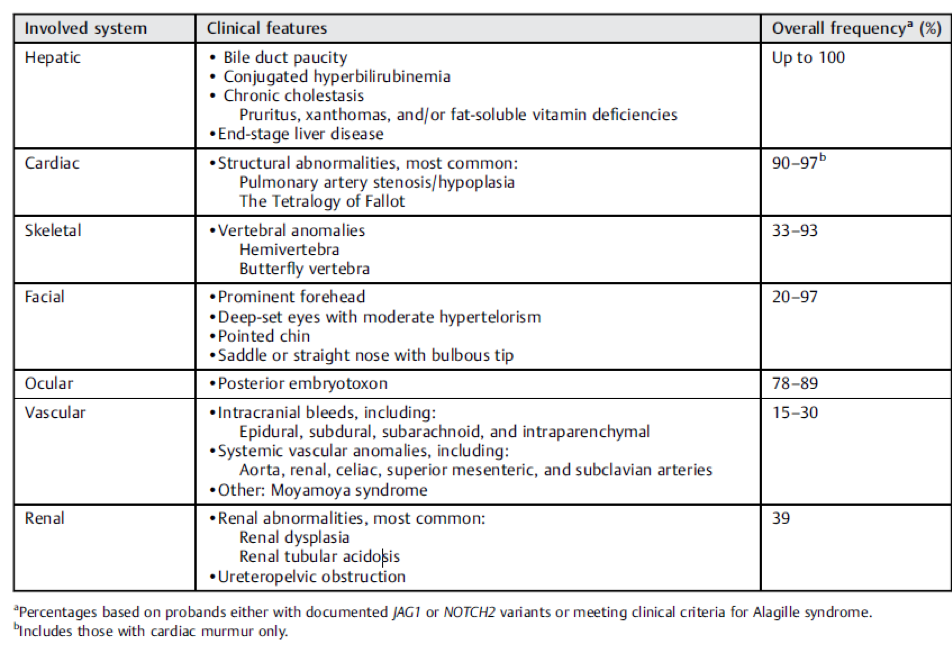
Taken from: Kohut, Taisa J., Melissa A. Gilbert, and Kathleen M. Loomes. "Alagille Syndrome: A Focused Review on Clinical Features, Genetics, and Treatment." Seminars in Liver Disease. Thieme Medical Publishers, Inc., 2021.
Diagnosis
Alagille syndrome should be suspected in all individuals with:
- Histologic findings of bile duct paucity on liver biopsy
- Three of the following five major clinical features:
- Cholestasis
- Cardiac defects
- Skeletal abnormalities
- Ophthalmologic abnormalities
- Characteristic facial features
In patients who meet clinical criteria, the diagnosis can be further confirmed by identifying a heterozygous pathogenic variant (JAG1 or NOTCH2) on molecular genetic testing. In individuals with an affected first-degree relative and a JAGGED1/NOTCH2 mutation, the presence of one or more clinical features is sufficient to make the diagnosis. The table below (from the ChiLDReN Network) provides a summary of diagnostic criteria based on family history, biopsy findings, and genetic testing results.
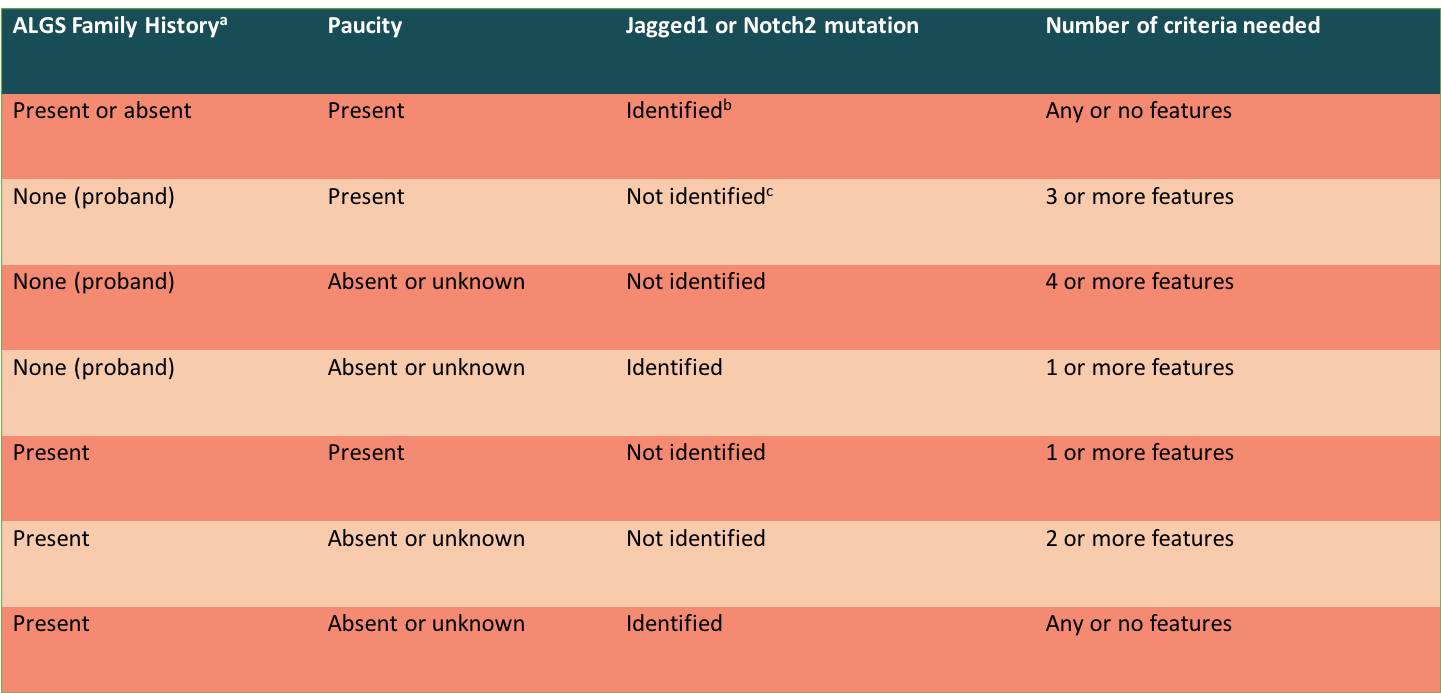
Taken from: The Child Liver Disease Research Network (https://childrennetwork.org/For-Physicians/Alagille-Syndrome-Information-for-Physicians) Of note, patients with Alagille syndrome who undergo Kasai procedure (hepatoportoenterostomy) have been shown to have no clinical benefit and worsened outcomes after surgery, including early transplantation and increased mortality. In patients with a non-diagnostic liver biopsy for biliary atresia, further assessment for extra-hepatic manifestations of Alagille syndrome may help guide diagnosis and appropriate management.