Biliary atresia

Case #1
Clinical History and Presentation:
- This patient is a 2-month-old full-term male with an unremarkable pregnancy and delivery who presented with ongoing jaundice, hyperbilirubinemia, and elevated transaminases.
- Laboratory studies were notable for Hgb 11, increased RDW, INR 1.6, albumin 3.6, K 5.3, AST 1028, ALT 354, alkaline phosphatase 723, total bilirubin 14.7, and direct bilirubin 7.12.
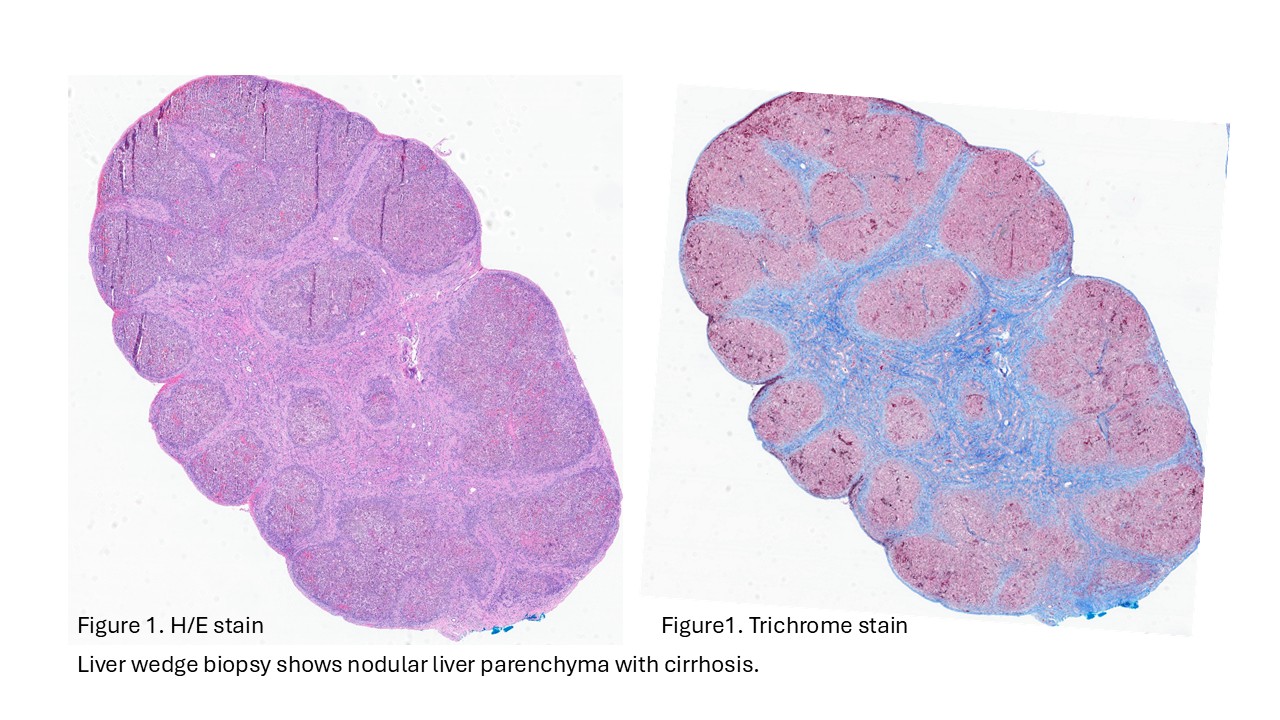
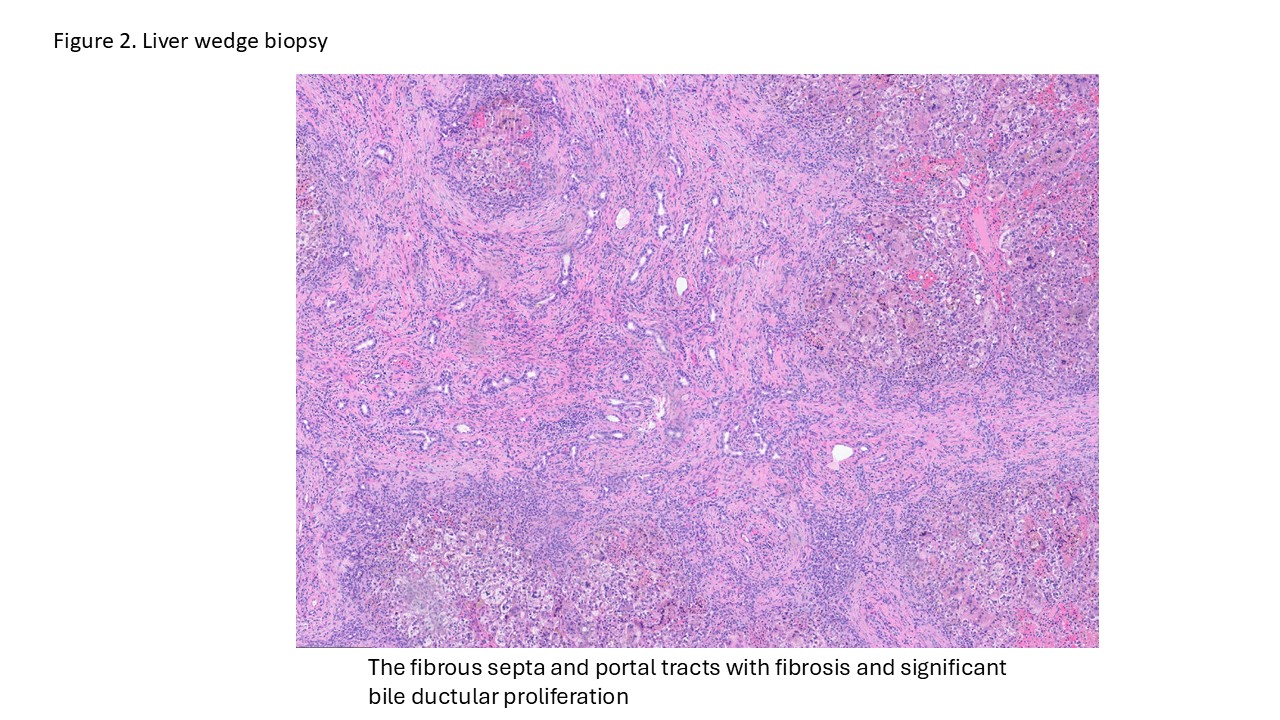
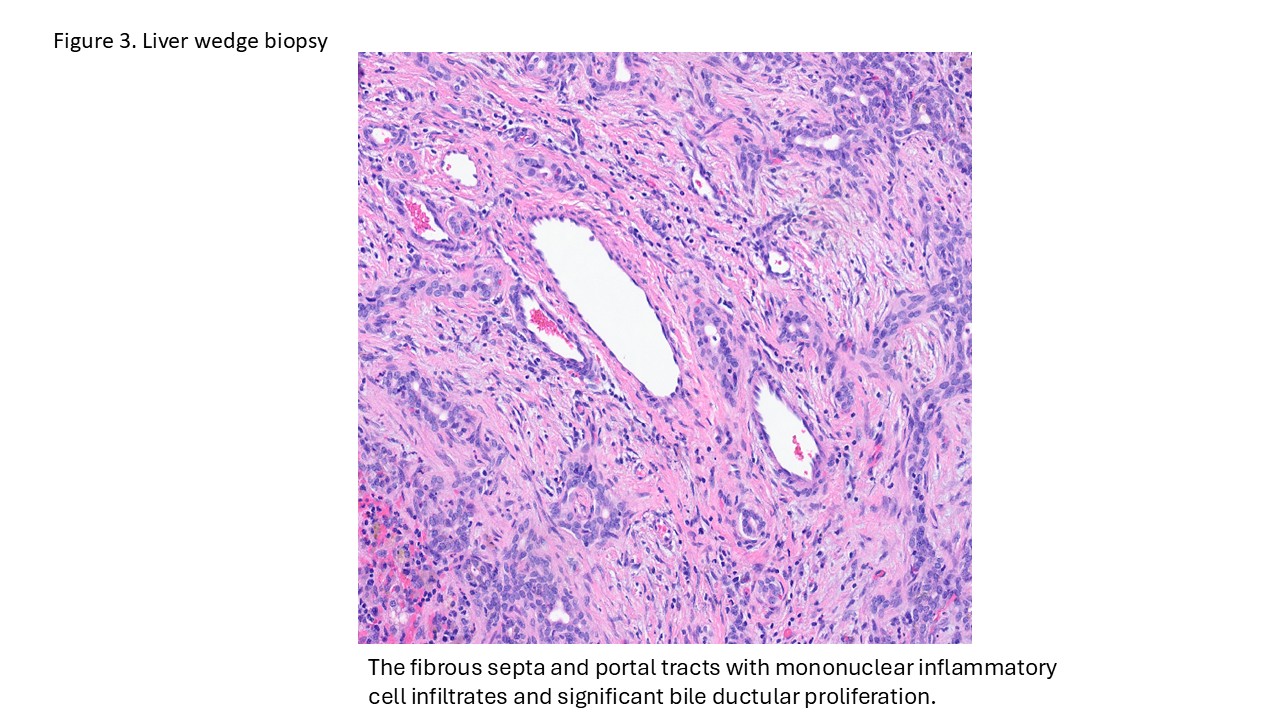
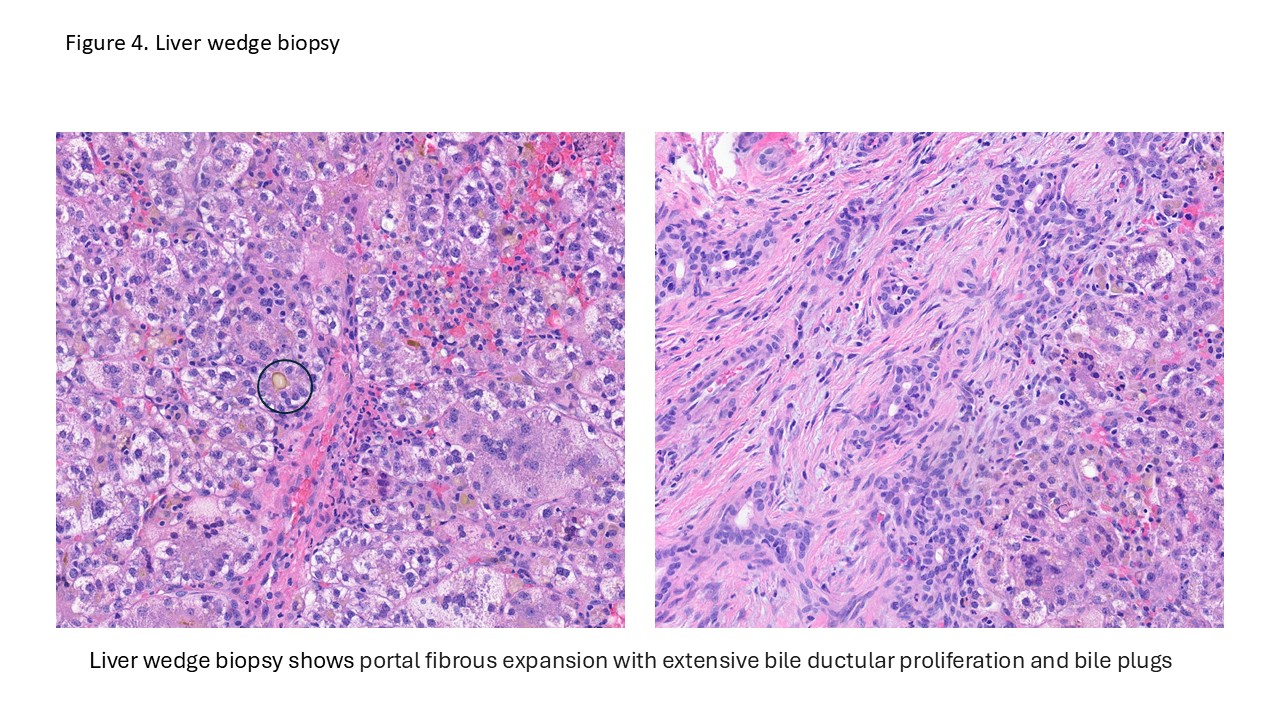
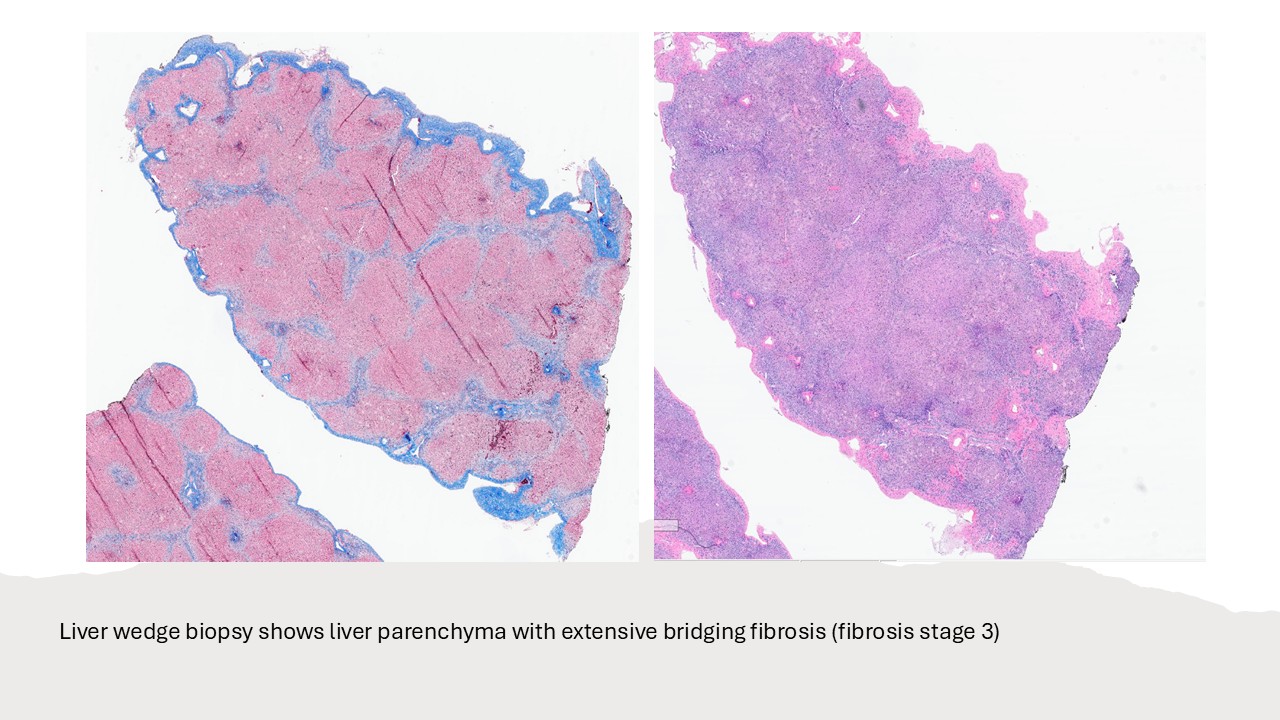
- Liver ultrasound on arrival demonstrated mild hepatomegaly with coarsened parenchymal texture, a possible nodular surface contour, and absence of the gallbladder. Labs showed worsening direct hyperbilirubinemia. The infant underwent wedge liver biopsy and cholangiogram, which were consistent with biliary atresia; consequently, a hepatic portoenterostomy was performed.
- In the operating room, the liver was noted to be scarred and nodular/cirrhotic. There was an atretic common bile duct and hepatic duct without a lumen, consistent with Type III biliary atresia.
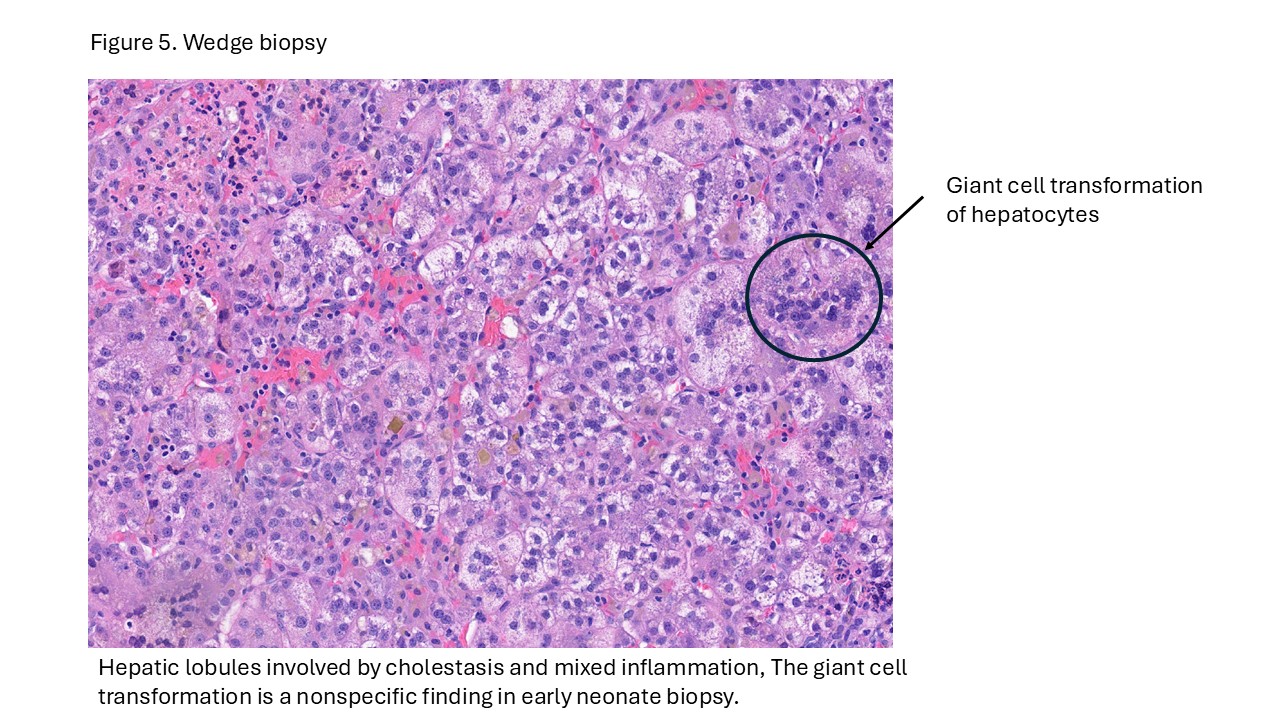
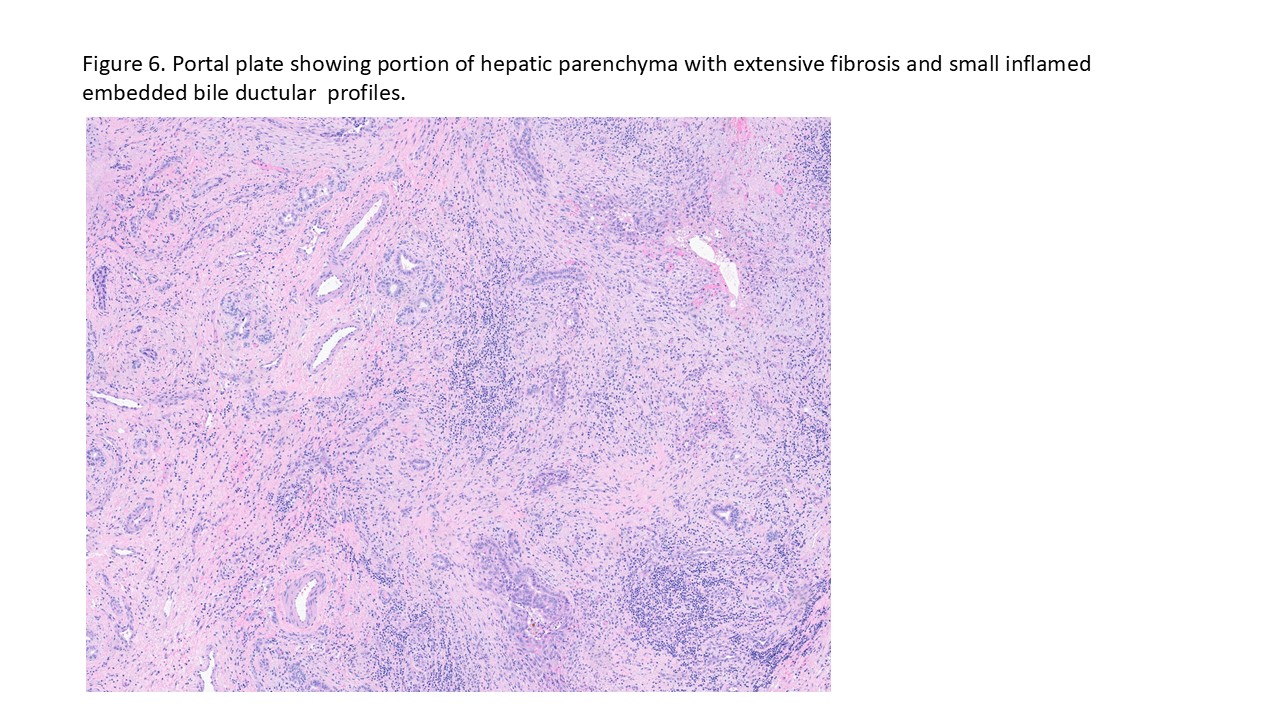
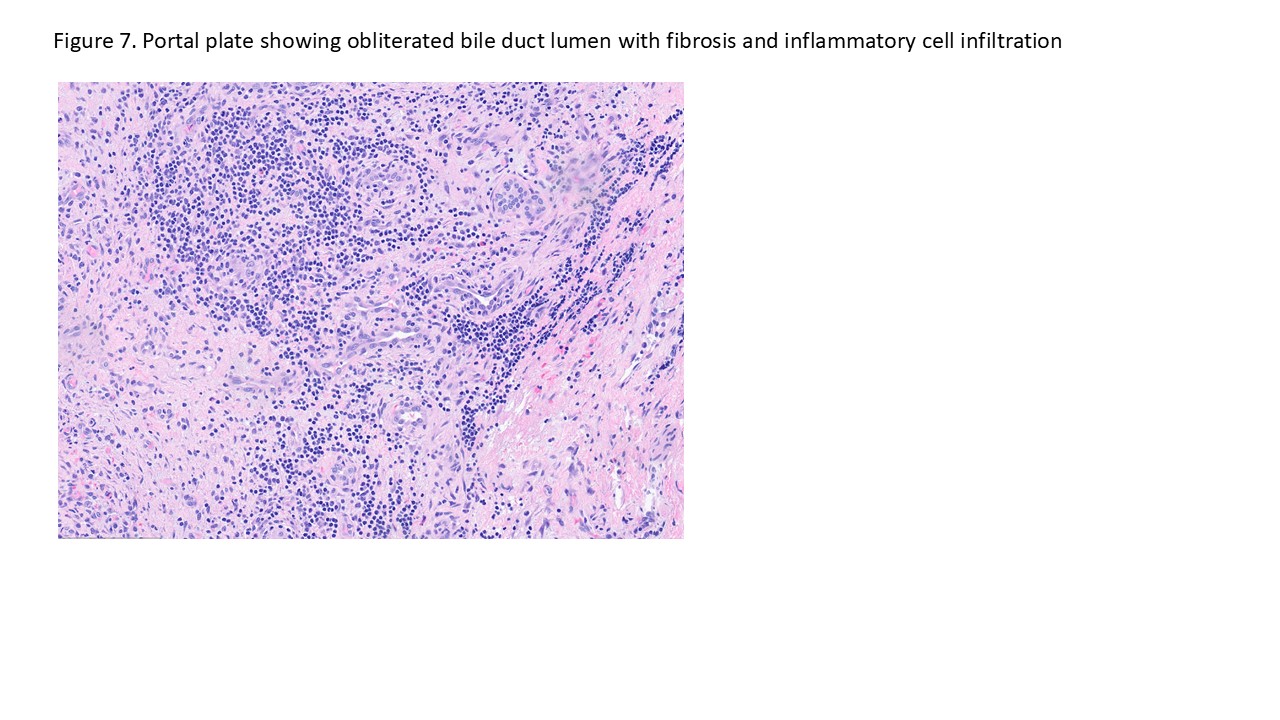
Case # 2
Clinical History and Presentation:
This 7-week-old infant with conjugated hyperbilirubinemia and light-colored stools undergoes exploratory laparotomy to evaluate for possible biliary atresia. The gallbladder appears as a scarred-down streak remnant with no evident lumen. The common bile duct and hepatic duct are also atretic. A cholangiogram is attempted, but no discernible lumen can be identified. A Kasai procedure is subsequently performed.
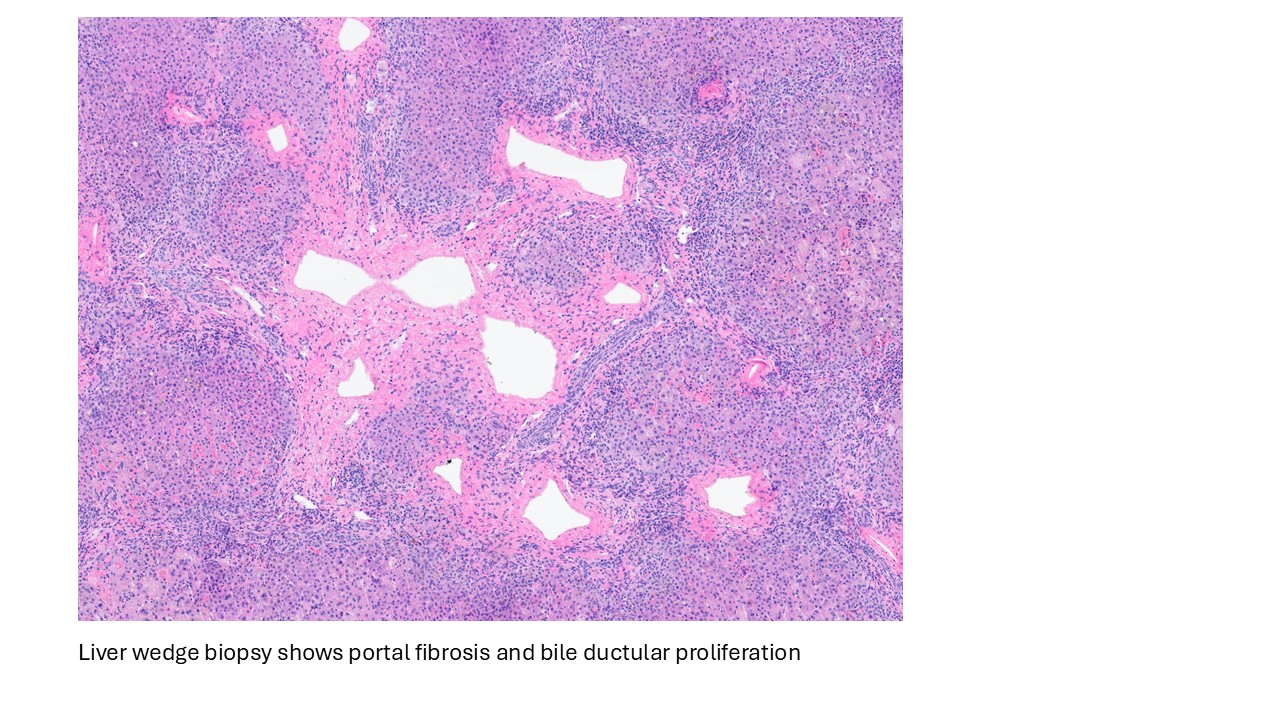
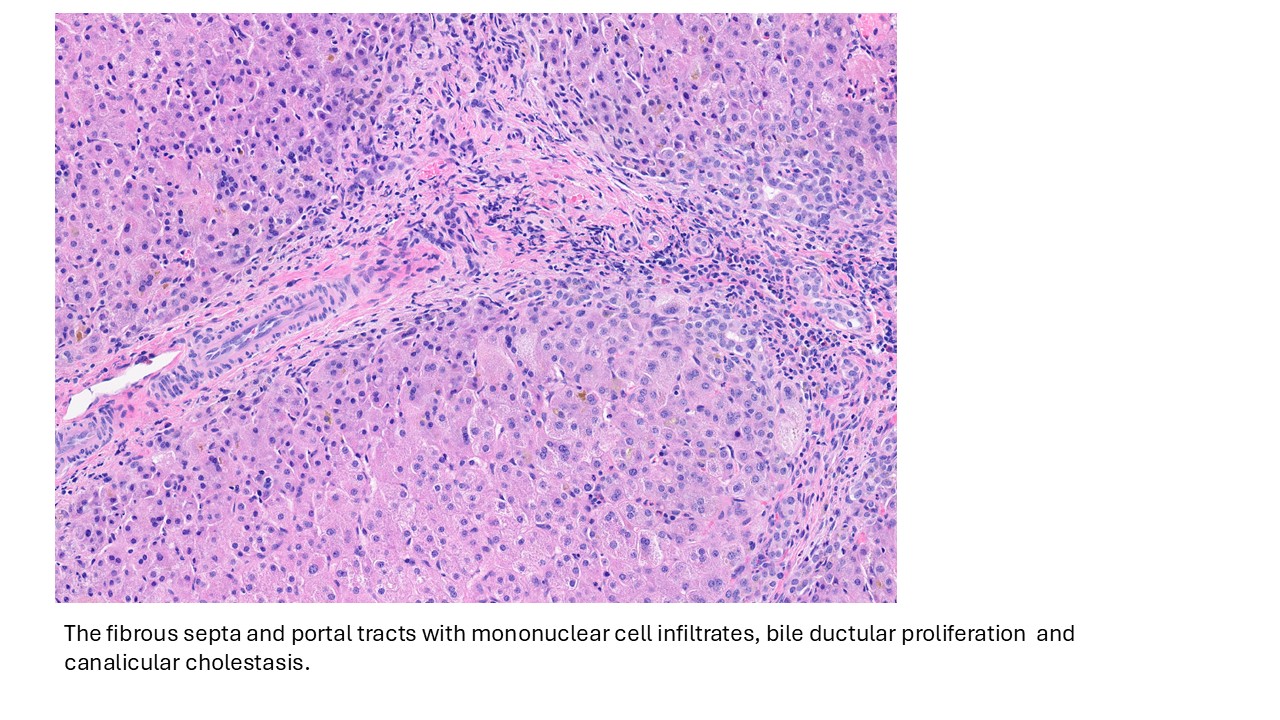
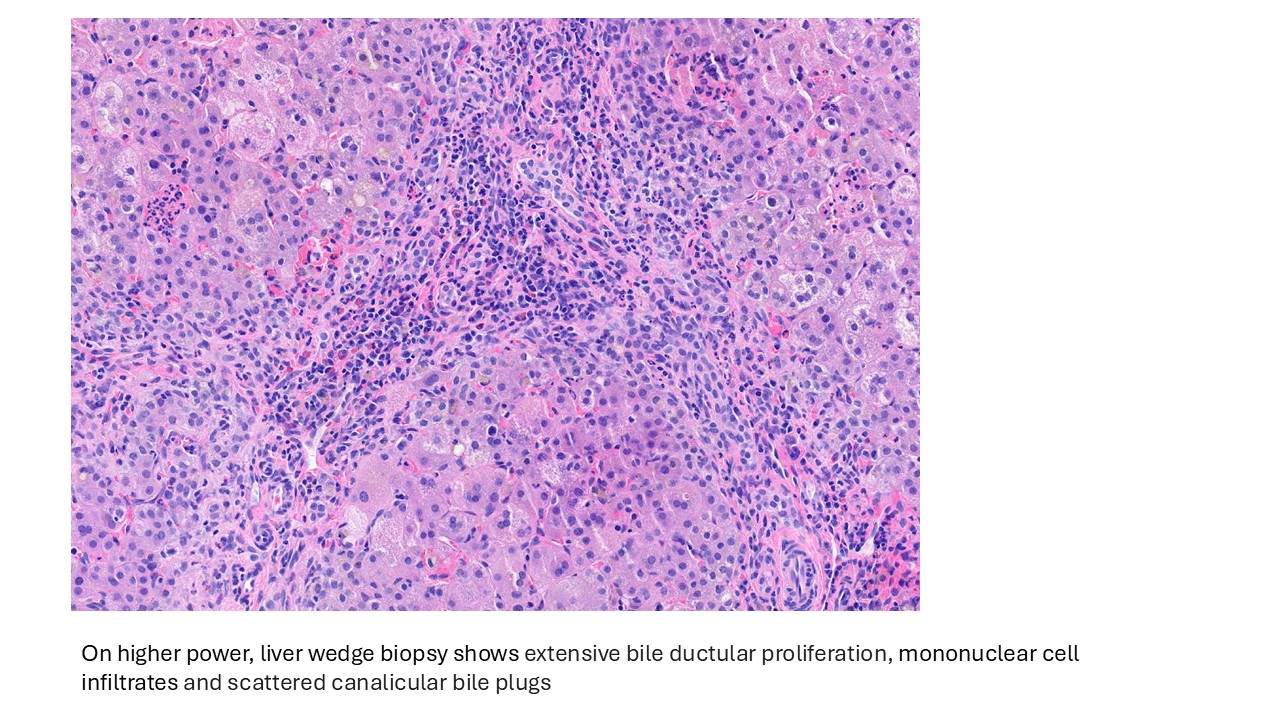
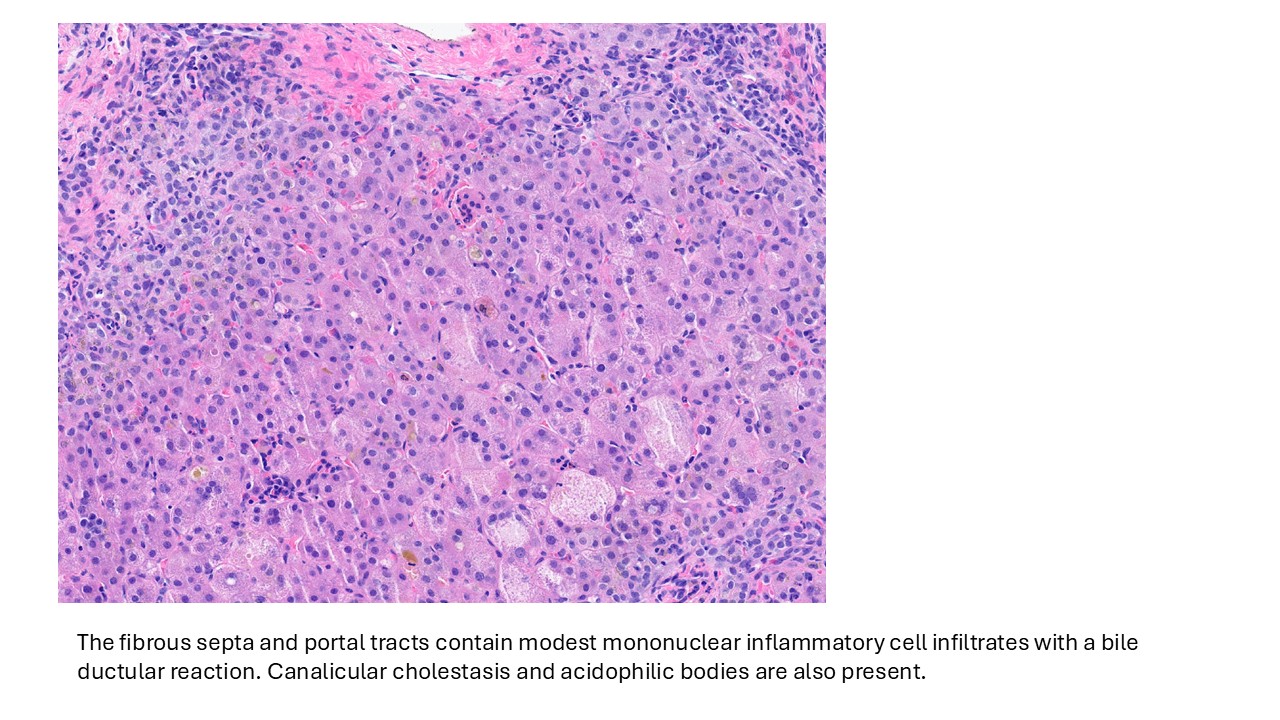
Discussion
Biliary atresia is a progressive fibro-obliterative cholangiopathy of infancy involving the extrahepatic and variable portions of the intrahepatic biliary tree, resulting in impaired bile flow, cholestasis, and eventual biliary cirrhosis if untreated. The pathogenesis is multifactorial and incompletely understood, with proposed mechanisms including abnormal embryologic bile duct development, immune-mediated bile duct injury, viral or environmental triggers, and genetic susceptibility leading to progressive inflammatory destruction of bile ducts. Clinically presents as cholestatic jaundice of infants with normal gestational age. Biliary atresia is classified into three types based on the level of biliary obstruction. Type 1, accounting for approximately 5% of cases, involves atresia of the common bile duct with preservation of the right and left hepatic ducts and common hepatic duct; the gallbladder is often hypoplastic but may be identifiable. Type 2, representing about 2% of cases, is characterized by atresia of the common hepatic duct, typically with an empty or poorly developed gallbladder. Type 3, the most common form comprising nearly 90% of cases, involves atresia at the level of the porta hepatis affecting the proximal extrahepatic biliary tree, often accompanied by an atretic gallbladder. Histologically, liver biopsies classically demonstrate portal tract expansion with bile ductular proliferation, bile plugs within ductules, portal edema, mixed inflammatory infiltrates, and progressive portal/periportal fibrosis; advanced cases may show bridging fibrosis and cirrhosis. Giant cell transformation and lobular cholestasis may also be present, particularly early in the disease course. The diagnosis of biliary atresia requires integration of clinical, laboratory, radiologic, and histopathologic findings. Infants typically present with persistent jaundice, acholic stools, dark urine, and conjugated hyperbilirubinemia within the first several weeks of life. Imaging studies, including abdominal ultrasound and hepatobiliary scintigraphy/Hepatobiliary iminodiacetic acid (HIDA) scan, may demonstrate absent or impaired bile flow and abnormalities of the extrahepatic biliary tree. Liver biopsy remains an important diagnostic tool, with characteristic findings including bile ductular proliferation, bile plugs, and portal/periportal fibrosis. Intraoperative cholangiography is considered the diagnostic gold standard and confirms the absence or discontinuity of the extrahepatic bile ducts. Management centers on early surgical intervention with Kasai portoenterostomy, ideally before 60 days of life, to restore bile drainage and improve native liver survival; however, many patients ultimately develop progressive liver disease and require liver transplantation later in life. Prognosis is strongly associated with age at Kasai procedure and postoperative bilirubin clearance, with earlier intervention correlating with improved long-term native liver survival.
Differential diagnosis
- Neonatal hepatitis: Typically shows prominent lobular disarray with lobular inflammation, multinucleated giant cell transformation, mild cholestasis, with less bile ductular proliferation and portal fibrosis compared with biliary atresia.
- Alagille syndrome: Characterized by paucity of interlobular bile ducts (bile duct loss in more than 50% of evaluable portal tracts), usually without the prominent ductular reaction seen in biliary atresia. It is associated with mutations in JAG1 or NOTCH2 in most cases. Patients may have other systemic manifestations involving the liver, heart, eyes, vertebrae, facial features, vascular system, and occasionally kidneys.
- Alpha-1 Antitrypsin Deficiency: May demonstrate neonatal cholestasis and portal fibrosis. Patients typically have decreased serum alpha-1 antitrypsin levels, and PAS/D-positive cytoplasmic globules may not be apparent in infants younger than 3 months.
- Progressive familial intrahepatic cholestasis type 3 (PFIC3): Often demonstrates diffuse hepatocellular cholestasis with occasional ductular cholestasis, portal fibrosis, and florid ductular proliferation. It is associated with mutations in the ABCB4 gene, which encodes MDR3. Age at onset varies from infancy to 20 years and should be correlated with imaging and genetic findings.
- Metabolic and syndromic cholestatic disorders: May mimic biliary atresia clinically but often lack the characteristic combination of marked ductular proliferation, bile plugs, and progressive portal-based fibrosis.
- Total parenteral nutrition associated cholestasis: Associated with a clinical history of prolonged total parenteral nutrition. Infants are usually premature and may have a history of necrotizing enterocolitis.
References
- Vij M, Rela M. Biliary atresia: pathology, etiology and pathogenesis. Future Sci OA. 2020 Mar 17;6(5):FSO466. doi: 10.2144/fsoa-2019-0153. PMID: 32518681.
- Dědič T, Jirsa M, Keil R, Rygl M, Šnajdauf J, Kotalová R. Alagille Syndrome Mimicking Biliary Atresia in Early Infancy. PLoS One. 2015 Nov 30;10(11):e0143939. doi: 10.1371/journal.pone.0143939. PMID: 26618708.
- Davenport M. Biliary atresia: clinical aspects. Semin Pediatr Surg. 2012 Aug;21(3):175-84. PMID: 22800970.
- Gunadi, Sirait DN, Budiarti LR, Paramita VMW, Fauzi AR, Ryantono F, Afandy D, Yoshuantari N, Rinonce HT, Makhmudi A. Histopathological findings for prediction of liver cirrhosis and survival in biliary atresia patients after Kasai procedure. Diagn Pathol. 2020 Jul 2;15(1):79. PMID: 32616059.
- Superina R, et al; Childhood Liver Disease Research and Education Network. The anatomic pattern of biliary atresia identified at time of Kasai hepatoportoenterostomy and early postoperative clearance of jaundice are significant predictors of transplant-free survival. Ann Surg. 2011 Oct;254(4):577-85. PMID: 21869674
- Dědič T, Jirsa M, Keil R, Rygl M, Šnajdauf J, Kotalová R. Alagille Syndrome Mimicking Biliary Atresia in Early Infancy. PLoS One. 2015 Nov 30;10(11):e0143939. doi: 10.1371/journal.pone.0143939. PMID: 26618708.
- Ayoub MD, Kamath BM. Alagille Syndrome: Diagnostic Challenges and Advances in Management. Diagnostics (Basel). 2020 Nov 6;10(11):907. doi: 10.3390/diagnostics10110907. PMID: 33172025
- Mitchell E, Gilbert M, Loomes KM. Alagille Syndrome. Clin Liver Dis. 2018 Nov;22(4):625-641. doi: 10.1016/j.cld.2018.06.001. Epub 2018 Aug 22. PMID: 30266153.
- Morotti RA, Suchy FJ, Magid MS. Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and 3: a review of the liver pathology findings. Semin Liver Dis. 2011 Feb;31(1):3-10. doi: 10.1055/s-0031-1272831. Epub 2011 Feb 22. PMID: 21344347.
Acknowledgements:
Dr. John A Hart