Why are patients with cirrhosis susceptible to hepatorenal syndrome (HRS)?

Acute kidney injury (AKI) occurs in approximately 20 % of hospitalized patients with cirrhosis and is associated with increased mortality. The most common causes of AKI in patients with cirrhosis include pre-renal, acute tubular necrosis (ATN) and hepatorenal syndrome (HRS). HRS has a very high mortality rate and the worst prognosis among the etiologies of AKI, with a survival rate of 35%.
The differentiation of these 3 main categories is critical, as the prognosis and treatments differ significantly based on etiology.
What is the history behind our understanding of HRS?
Coexistence of liver/kidney damage was highlighted as early as 1827 by Richard Bright. He describes 7 cases of dropsy (edema) due to liver disease. In his case reports, he described the urine as diminished before death and the autopsy as “rather pale, with irregular vascularity but in structure normal”!
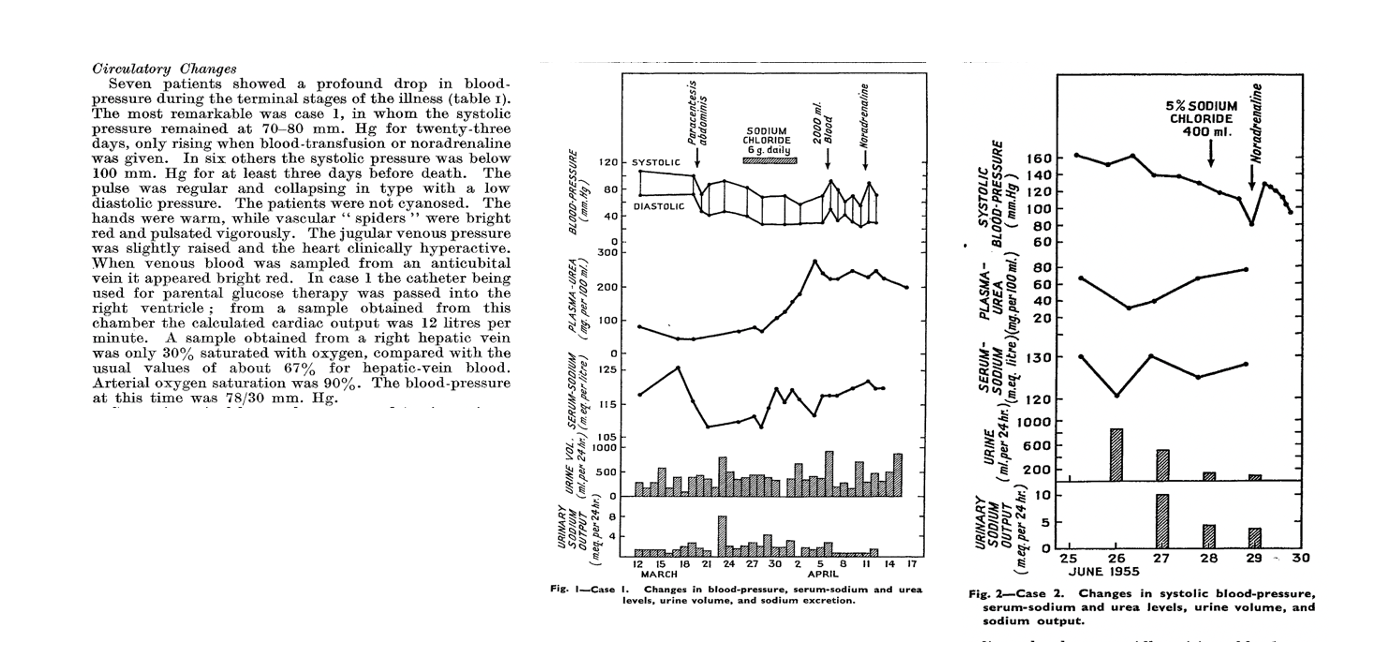
However one of the first studies to shed light on HRS’ pathogenesis is a study by Hecker and Sherlock published in Lancet in 1956, which suggested that peripheral arterial vasodilation plays a major role. The study describes the detailed hemodynamic assessment and hospital course of 9 patients with cirrhosis or subacute hepatic failure, ascites, hypotension, and hyponatremia who developed progressive azotemia. They observed significant hypotension, high cardiac output, and warm shock, consistent with profound vasodilation and hyperdynamic circulation. They did not observe any evidence of structural kidney damage on evaluation of the urine microscopy or autopsy postmortem. In four cases, therapeutic paracentesis, and in one case, bacterial infection, was cited as the precipitating event. The study describes the unifying syndrome as patients with “advanced liver disease, ascites, who have deepening jaundice, a falling serum sodium level and urine output and a systolic pressure below 100 mmHg with fatal outcome”. This is the first study that recommends the use of norepinephrine and volume expansion with blood transfusions for the management of HRS.
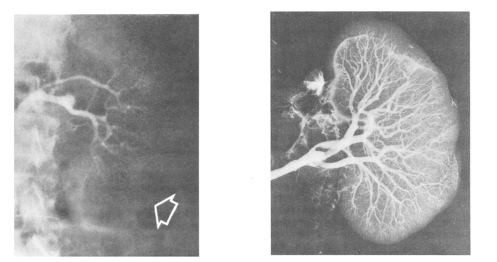
This is a grim but fascinating picture from a study from 1970 that shows the extreme vasoconstriction in the kidney of a patient with HRS (left panel). The picture to the right is the angiogram of the same kidney performed postmortem which shows intact vasculature inside the kidney. Hepatorenal syndrome was thought to be a “functional” renal dysfunction that occurs in patients with decompensated cirrhosis and ascites. Traditionally it was believed that there is no structural component and if the kidneys from someone with HRS are transplanted into another individual, they will start working just fine.
What is the pathophysiology of HRS?
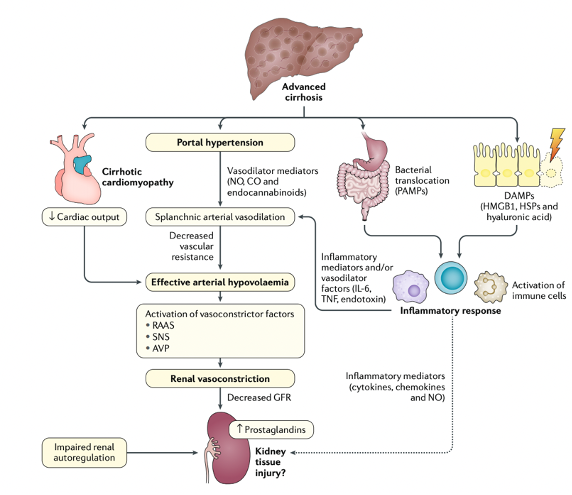
We know that portal hypertension results in splanchnic vasodilation, sequestration of the blood in the splanchnic circulation, and subsequent decrease in effective arterial blood volume. This decrease in effective volume in turn results in activation of renin-angiotensin-aldosterone and sympathetic nervous systems as compensatory mechanisms. The effect of these compensatory mechanisms is an increase in water and sodium retention from the kidneys, which leads to refractory ascites. Simultaneously, the increase in renal vasoconstriction and reduction in renal perfusion can lead to a functional renal failure, A.K.A. hepatorenal syndrome.
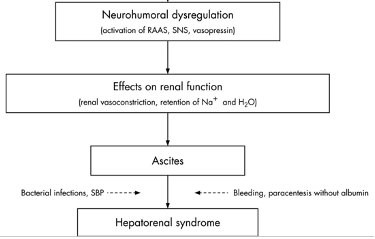
However, the traditional belief that HRS is purely a “functional” kidney dysfunction in the setting of hemodynamic changes caused by advanced portal hypertension, has recently been challenged with new research showing the role of systemic inflammation, oxidative stress and bile-related tubular damage in its development as additional “structural” components. One diagram below demonstrates different mechanisms involved in the development of HRS. The deterioration of systemic circulatory dysfunction is often triggered by a precipitating event such as bacterial infection (mainly spontaneous bacterial peritonitis), bleeding, NSAID use or paracentesis without albumin (second diagram below).
Why was the definition of HRS changed?
In 2007, the International Club of Ascites (ICA) classified HRS into types 1 and 2 (HRS-1 and HRS-2). HRS type one was defined as a rapidly progressive renal failure in the context of an acute event with an increase in serum creatinine to greater than 2.5 mg/dL within two weeks. HRS type 2 was defined as a slow progressive increase of creatinine to greater than 1.5 mg/dL, usually in the setting of refractory ascites.
However, this definition had many limitations. First, serum creatinine underestimates the real renal function in patients with cirrhosis due to muscle wasting, and a potential dilution of creatinine in the setting of an increased volume of distribution. Second, this strict threshold resulted in late diagnosis of this condition and would not allow for timely and proactive interventions to halt the progression.
For these reasons, in 2015, the ICA proposed new criteria for defining HRS. The new criteria adopted the AKI definitions used by the Kidney Disease Improving Global Outcomes (KDIGO) guidelines. Based on KDIGO, AKI is defined as an increase in Cr by ≥ 0.3 mg/dl within 48 hours or ≥ 1.5x baseline within 7 days. Furthermore, IAC proposed that if a baseline value within the previous 7 days is not available, the most recent value within the last 3 months can be used.


Despite the definition and the above diagnostic criteria proposed by ICA, the diagnosis of HRS remains challenging in clinical practice.
The true gold standard remains kidney biopsy, but it is rarely (if ever) done in these patients with decompensated cirrhosis given concern for increased risk of bleeding. At this point a clinical decision making incorporating the patient’s phenotype (ascites (usually refractory), hyponatremia, low MAP), and ruling out other etiologies (pre-renal and ATN) remain our best practical means of differentiating these etiologies.
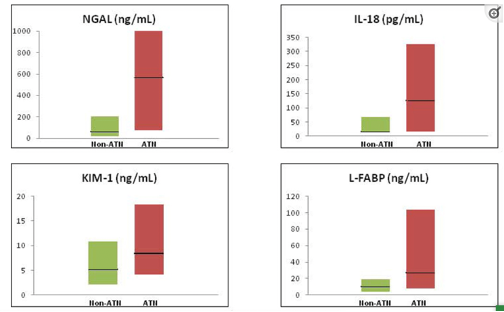
There has been a growing interest in novel kidney injury biomarkers that have the potential to differentiate between ATN and other causes of AKI in cirrhosis. Urinary neutrophil gelatinase-associated lipocalin (NGAL) has demonstrated great performance, and cut offs for NGAL have been proposed to assist in differentiation of ATN vs HRS. Other potential markers are illustrated below. Unfortunately, despite their utility, they are not widely available in clinical practice, and their use have remained mainly limited to the research setting.
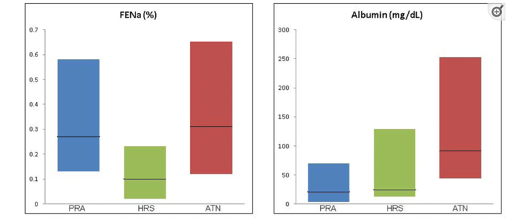
The utility of FeNa in patients with cirrhosis and AKI is often dismissed as it is <1% regardless of etiology (structural vs functional); however FeNa appears to be lowest in patients with HRS (<0.1%) given the extreme sodium avid state in these patients.
Another useful diagnostic tool which can assist in differentiating the etiologies of AKI is point-of-care echocardiography (POCE) and measurement of inferior vena cava (IVC) diameter and collapsibility to assess intravascular volume status. A pilot study in patients with cirrhosis and AKI who were assigned a clinical diagnosis of HRS type one, revealed marked heterogeneity. Among the 53 patients evaluated by POCE, 23% were reclassified as volume depleted, 21% were reclassified as volume overloaded, and 15% were reclassified as having intra-abdominal hypertension (IAH). The study highlights that volume status varies significantly among patients labeled as having HRS-1 and that the therapeutic interventions should be tailored to each patient’s volume status. One size does not fit all, although albumin resuscitation is a great choice for a patient who is still intravascularly volume depleted, it can potentially cause serious adverse events such as pulmonary edema and respiratory failure in a patient that is already volume overloaded.
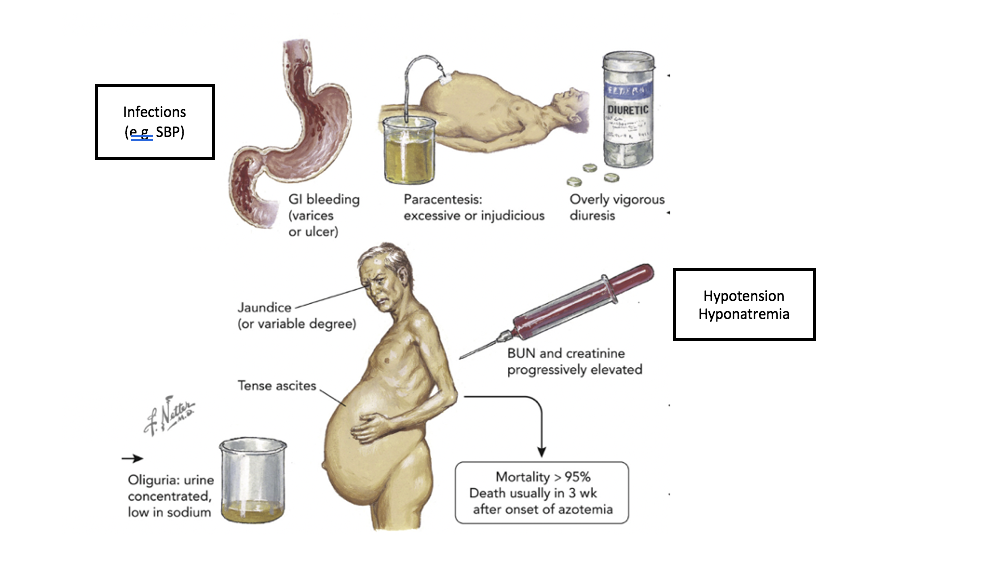
In summary, there is great interest and growing research in better markers and tests to more objectively diagnose HRS. However, in the absence of a simple high performing diagnostic test, the best tool remains our clinical judgement incorporating different aspects of the history, physical, laboratory values and criteria proposed by ICA. Here is a great illustration showing the phenotype of a patient with HRS and precipitating events. (Picture is adapted from Garabed et al). The 2 black boxes are added for this review. The original colored figure in the center was painted by Frank Netter 1985.
Why and how do we treat HRS?
Given the main pathophysiology of HRS is driven by splanchnic and systemic vasodilation,
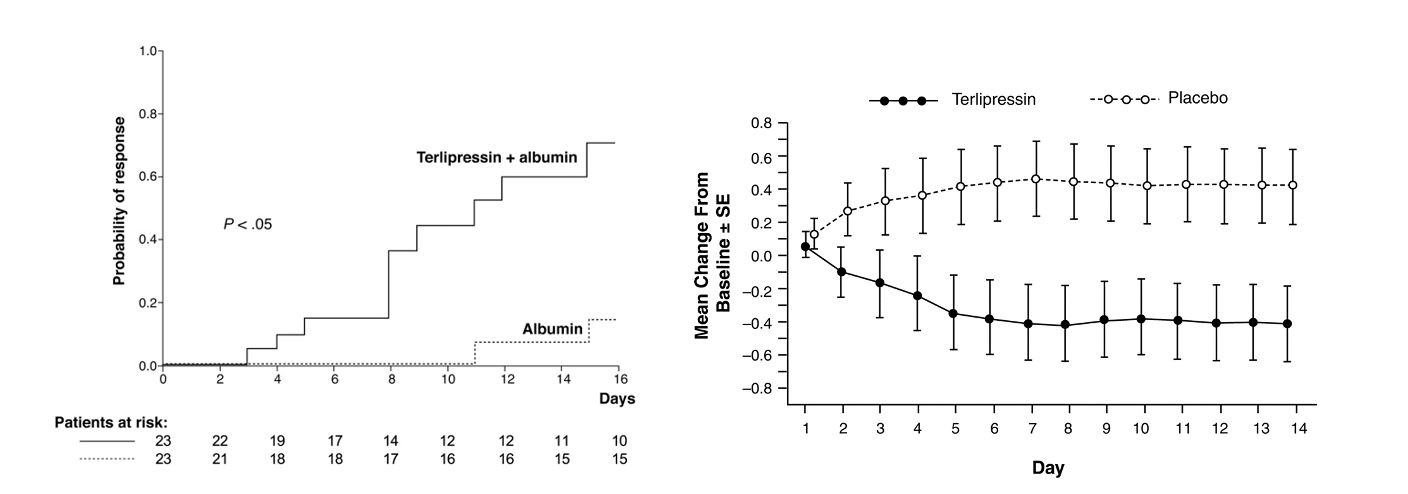
current treatments for HRS focus on increasing the effective arterial circulation with vasoconstriction and an increase in intravascular volume. The standard of care in Europe is terlipressin (vasopressin analog) and albumin which has the most evidence in terms of its efficacy to improve renal function; however, there is inconsistent evidence in terms of effect on survival. What all studies have in common, is the demonstration that patients with a resolution of HRS have significantly better survival compared to the ones that don’t.
The graph on the left panel shows the results of terlipressin plus Albumin compared to albumin alone in improving kidney function. The right panel shows the mean change in serum creatinine compared between terlipressin plus Albumin vs albumin alone in an RCT of 112 patients. However they were not able to show a difference in survival between the terlipressin versus the placebo group.
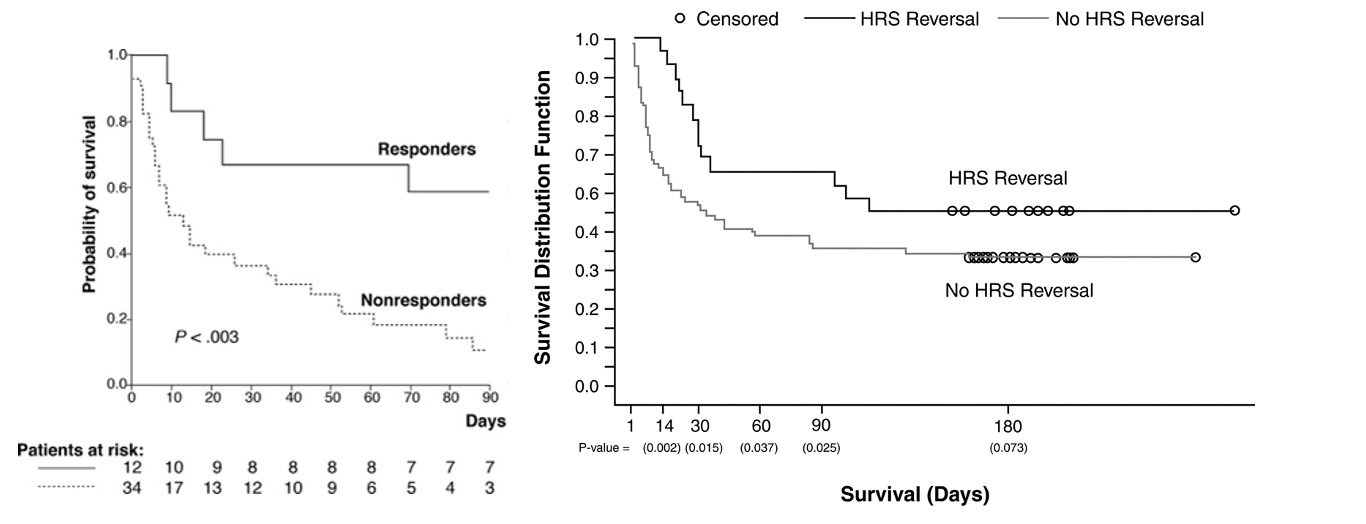
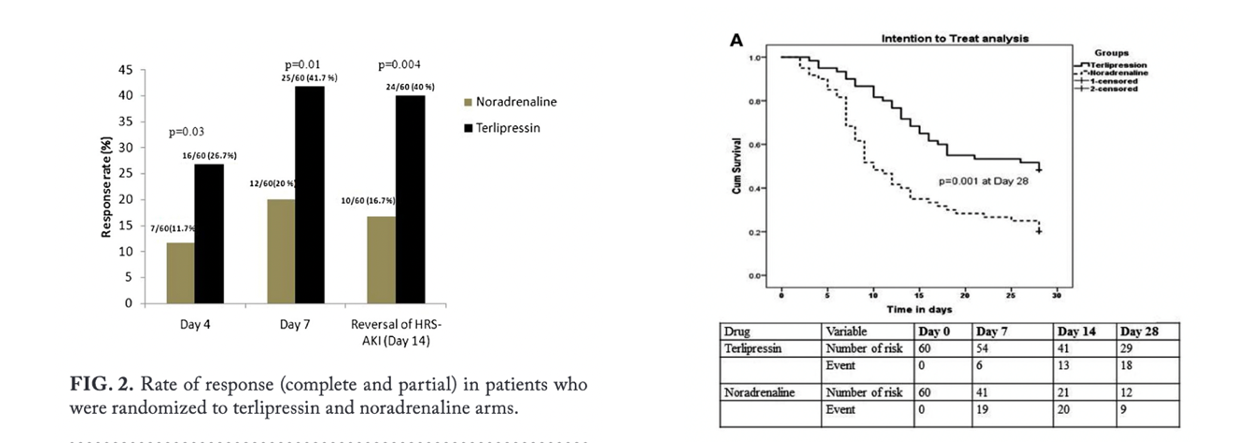
Here are the graphs from the above trials showing the significant difference in survival based on response (reversal of HRS) vs lack of response.
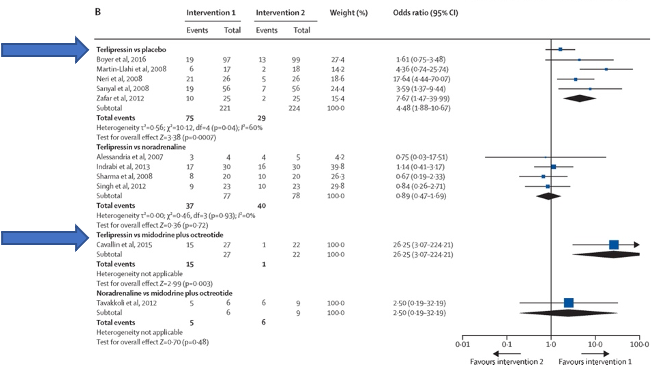
A systematic review of 13 randomized trials (739 patients) with HRS type one, showed improvement in reversal of HRS with terlipressin as compared with placebo or midodrine and octreotide. None of the other pairwise comparisons showed any difference. Another Cochrane meta-analysis of 25 trials (1263 participants) including both HRS type one and HRS type two did not show any evidence of a difference in mortality between the different interventions. However it also showed that recovery from hepatorenal syndrome was higher with terlipressin compared to octreotide/midodrine or placebo.
CONFIRM trial is the most recent randomized trial of 300 patients in North America. It was designed to look at the primary end point of HRS reversal in patients with HRS-type one. The study achieved its primary end point of verified HRS reversal (32% in terlipressin group vs 17% in placebo group, p:0.006), however there were more adverse events including death from respiratory failure in 11% of patients in the terlipressin group and 2% in the placebo group which resulted in some safety concerns, and as such this treatment has not yet been approved by the FDA.
Currently, given unavailability of terlipressin in the United States, many centers treat patients with HRS with octreotide and midodrine, and if there is no response, these patients can be considered for norepinephrine to maintain MAPS about 10-15 mmHg higher than baseline. The data on the efficacy of norepinephrine as compared to terlipressin is mixed. Two open label trials showed similar efficacy and safety profile when compared to terlipressin. However a recent randomized trial showed terlipressin to be more effective than norepinephrine in patients with Acute on Chronic Liver Failure (ACLF). The study used the new definition of HRS (i.e. HRS-AKI).
Although in most centers norepinephrine is limited to intensive care units, a study by Kwong et al showed that it can be safely used outside of intensive care setting in patients with HRS.
Despite the response to treatment, patients remain at risk for new episodes of HRS-AKI after withdrawal of treatment. Liver transplant remains the ultimate treatment for patients with HRS-AKI regardless of their response to therapy. However, it is often challenging to ascertain how much renal recovery is possible after liver transplantation, and when should patients be listed for Simultaneous Liver Kidney Transplant (SLK). In United States patients who have suffered from AKI for ≥ 6 weeks are thought to be unlikely to recover renal function after liver transplant, and are therefore eligible to be listed for SLK.
Since liver transplantation is limited by organ availability and strict eligibility criteria, some studies have explored the role of TIPS in the management of HRS. TIPS acts upstream in the pathophysiology of HRS by decreasing the portal pressure and thus shunting the blood that has been sequestered in the splanchnic circulation to the systemic circulation, thereby improving the effective arterial volume. Small observational studies have shown promising outcomes with TIPS in patients with HRS.
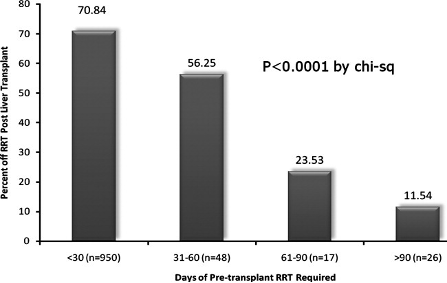
In conclusion, management of AKI in patients with decompensated cirrhosis remains a challenge. Our limited understanding of the extensive mechanisms that play a role in the pathogenesis of HRS and lack of markers differentiating between pre-renal, ATN, and HRS has been the main barrier in terms of advancing treatments for this disease. It is likely that patients participating in our current trials for management as HRS- type one actually have ATN, and are misclassified as having HRS. This could explain why 50% of patients do not respond to terlipressin. Our inability to accurately classify patients with HRS vs ATN can be seen in data available on patients with extended duration of RRT prior to liver transplant. Although currently GFR <30 or HD for 6-8 weeks qualifies patients for SLK, in a cohort of such patients who were only transplanted for liver, 24% of patients who were on HD for 8-12 weeks, and 11% of patients who were on HD for more than 12 weeks had renal recovery post-transplant.