Why is Nutrition So Important in Alcohol Associated Liver Disease?

Malnutrition is common in alcohol associated liver disease (ALD). Malnutrition, as defined by WHO, refers to deficiencies, excesses, or imbalances in a person’s intake of energy and/or nutrients. In the 2021 AASLD guidance on malnutrition, frailty, and sarcopenia in patients with cirrhosis, malnutrition is defined as “a clinical syndrome that results from deficiencies or excesses of nutrient intake, imbalance of essential nutrients, or impaired nutrient use,” with an operational definition of an “imbalance (deficiency or excess) of nutrients that causes measurable adverse effects on tissue/body form (body shape, size, composition) or function and/or clinical outcome”. It is important to note that malnutrition represents a variety of nutritional disorders that encompasses a range of BMIs, including patients who are overweight or obese.
As briefly explored in my last Why? Series post, it was not until the 1960s that alcohol was identified as a causative agent of liver disease. Prior to this discovery, it was actually a common belief that the liver damage seen in those with heavy alcohol use was instead, from malnutrition. Given the widespread early belief that liver disease associated with heavy alcohol use was in fact due to malnutrition, nutrition was among the first proposed treatments of alcohol associated liver disease. However, there was, and still is, quite a debate regarding the composition of the ideal diet for this population. We will explore this controversy in more detail below.
How common is malnutrition in patients with ALD?
The answer is…very common! A 1984 study of 363 US veterans set out to characterize malnutrition in patients with alcohol associated hepatitis (AAH). The authors evaluated nutritional status using parameters suggested in Blackburn et al.’s 1977 paper on nutritional assessment in the hospitalized patient. The following figure from Blackburn summarizes the major parameters assessed, with attention to surrogate markers for the components of body composition. These parameters included an assessment of total body fat through measurement of the triceps skin fold, assessment of visceral protein status through transferrin and TIBC, immune function through total lymphocyte count, and assessment of lean body mass through creatinine-height index.
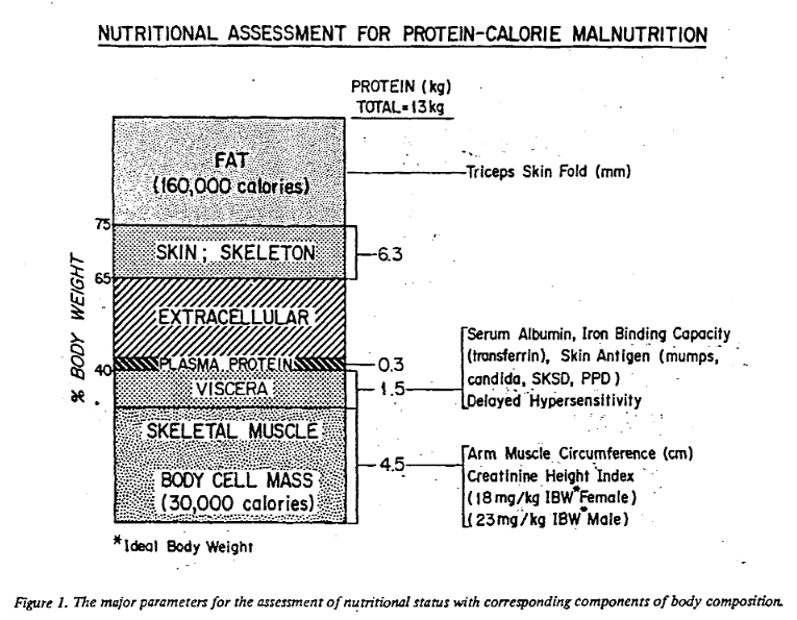
They also identified marasmus-like and kwashiorkor-like malnutrition. Kwashiorkor-like malnutrition was defined by the presence of low circulating visceral proteins (albumin, retinol-binding protein, and transferrin) and a suppressed immune response, which was defined by lymphocytopenia and delayed hypersensitivity as measured through a lack of response to a battery of skin tests including reaction to dinitrochlorobenzene (DNCB), which is a substance used to treat warts. Marasmus was diagnosed with the presence of decreased lean body mass (estimated by creatinine-height index), loss of stored fat (estimated by triceps skinfold thickness), and a lack of response to skin tests plus DNCB. The following table from Blackburn provides the parameters used to diagnose patients with kwashiorkor and marasmus-like malnutrition.
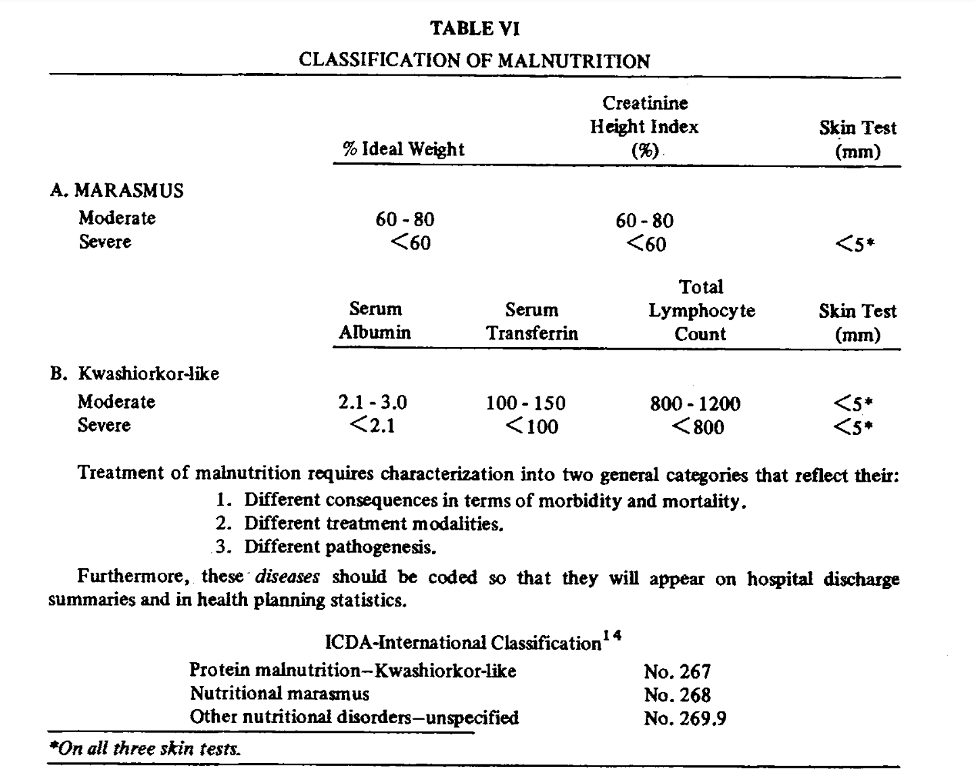
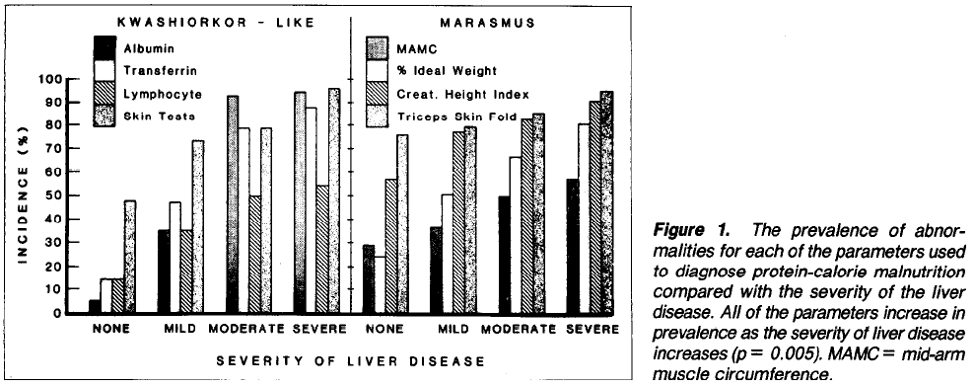
This VA study found that all patients with liver disease had some evidence of malnutrition, with some patients having features of kwashiorkor or marasmus, or both. They found that the incidence of malnutrition increased with increased severity of liver disease. The figure below illustrates the malnutrition-pattern and the increasing incidence of malnutrition with liver disease severity.
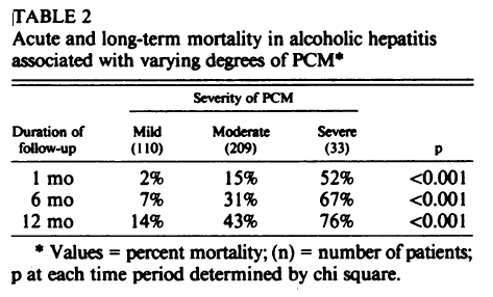
Another 1986 study by the same authors that included 352 hospitalized veterans with AAH, specifically looked at protein calorie malnutrition, using the same Blackburn criteria. They found increased 30-day mortality in patients with severe versus mild protein calorie malnutrition, with mortality of 52% and 2%, respectively, illustrated in the table below. They also found that as the severity of protein calorie malnutrition increased, the severity of AAH increased as did the presence of ascites and hepatic encephalopathy.
More recent studies on patients with cirrhosis due to alcohol undergoing transplant evaluation found that 84% were malnourished, despite 68% being overweight or obese. This study highlights that BMI is not always useful in determining nutritional status, as it does not capture sarcopenia and micronutrient deficiencies. Interestingly, this study did not find significant differences in 1-year survival between malnourished and well-nourished patients. The authors, however, did find that patients with malnutrition had significantly longer length of stays both at the time of listing and post-transplant.
How does alcohol use impact nutrition?
Alcohol use impacts nutrition via several mechanisms. First, alcohol is a high calorie substance, with ~7kCal/g, compared to 4 kCal/g in carbohydrates and 9 kCal/g in fat. With the high energy content per gram of alcohol, heavy alcohol consumption can displace calories from more nutritious sources. Studies have shown that moderate alcohol intake, defined as consuming 16% or less of total calories from alcohol, is associated with slightly increased total energy intake, with these alcohol often displacing calories from carbohydrates in the diet. In patients who consume more than 30% of their total calories from alcohol, intake of proteins and fats, as well as carbohydrates, is significantly reduced. In fact, individuals with heavy alcohol use can consume the majority of their calories from alcohol, leading to multiple nutritional deficiencies. Alcohol also has toxic effects on the gastrointestinal tract that leads to impaired digestion and absorption. In fact, it has been shown to shorten the villi in the small bowel, resulting in reduced surface area for nutrient absorption.
Alcohol has additionally been shown to alter the gut microbiome with a shift towards firmicutes. This results in reduced gut metabolism of bile acids with decreased conjugated salts that can lead to impaired absorption of fat and fat-soluble vitamins. Furthermore, some patients with prolonged, heavy alcohol use develop chronic pancreatitis and exocrine insufficiency resulting in impaired digestion of macronutrients. When portal hypertension develops in either the setting of AAH or cirrhosis, nutrients bypass the liver without undergoing metabolic processing. This leads to less glycogen storage and early activation of gluconeogenesis between meals, leading to increased skeletal muscle proteolysis in order to provide amino acids for glucose synthesis.
Furthermore, patients with portal hypertension or cirrhosis frequently develop early satiety and anorexia. Patients with chronic liver disease also have higher prevalence of small intestinal bacterial overgrowth, which can lead to symptoms of bloating and worsen absorption of nutrients. Some of the micronutrient deficiencies, which we will discuss below, can also lead to dysgeusia. Social factors including food insecurity and access to nutritious foods also contributes to malnutrition in this population.
What micronutrients are most deficient and are these deficiencies causal?
Folic Acid
Folic acid deficiency is a well-known and frequent consequence of alcohol-use disorder, with folate deficiency evident in as many as 80% of hospitalized patients with AUD. This B-vitamin is required for the conversion of homocysteine to methionine (with B-12 serving as a co-factor), which is then converted to S-adenosylmethionine (SAM), which is the methyl donor for all DNA methylation reactions. Glutathione, a key antioxidant, is produced from homocysteine through the transsulfuration pathway, with both SAM and Vitamin B6 required for this process, as seen in the figure below. Deficiency of folic acid therefore can promote oxidative stress.
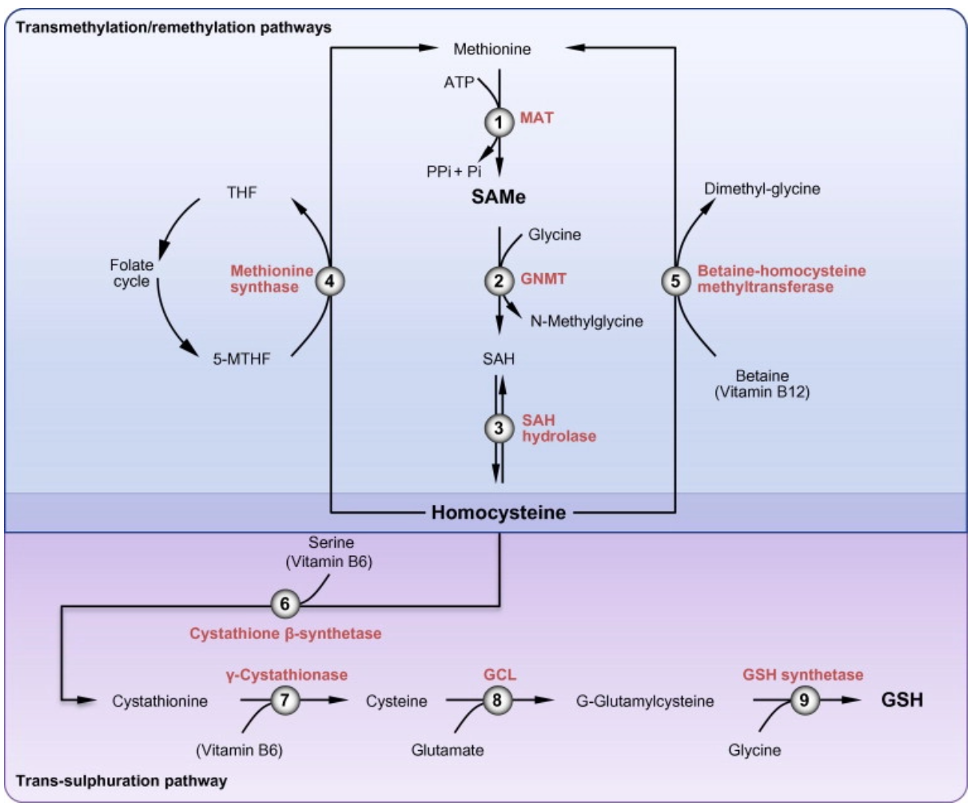
Do folic acid or B-vitamin deficiency play a role in the pathogenesis of ALD? A study in micropigs demonstrated that folic acid deficiency increases alcohol related injury through activation of CYP 2E1 and increased endoplasmic reticulum stress. Another animal study demonstrated that supplementing the diet with folate and B12 can improve liver injury caused by ethanol in rats, possibly by normalizing the plasma homocysteine levels. In a mouse study on NASH and hyperhomocysteinemia, the authors found that supplementation with vitamin B-12 and folate decreased hyperhomocysteinemia, restored autophagy, and improved hepatic histology in a mouse-model of pre-established NASH. These studies imply that B-12/folate deficiency and hyperhomocysteneimia are involved in the development of liver disease and may be treatment targets. Studies in humans, however, have demonstrated no benefit of SAM supplementation on liver-related outcomes.
Thiamine
Thiamine deficiency affects 80% of patients with alcohol use disorder. Thiamine deficiency in heavy alcohol use is due to a variety of mechanisms. Poor eating habits can lead to in adequate intake of dietary thiamine. Thiamine is absorbed in the small intestine, and ethanol can impair the intestinal absorption of thiamine by reducing thiamine entry into the enterocyte. The liver is the primary site of thiamine storage, with rat studies demonstrating that ethanol both causes the release of thiamine from the liver as well as inhibits thiamine uptake into hepatocytes. As thiamine is an important co-factor in cellular-metabolism, especially in the brain, thiamine deficiency can cause devastating consequences such as Wernicke’s encephalopathy and Korsakoff psychosis.
Vitamin D
Vitamin D deficiency is very common in patients with chronic liver disease, with 86.3% deficiency in those with cirrhosis and 49% in those without cirrhosis. Vitamin D has been shown to have antioxidant effects. Studies in rats have demonstrated that vitamin D deficiency exaggerates alcohol-related oxidative stress and liver injury, while vitamin D supplementation is protective against this same process. Vitamin D has also been shown to protect against alcohol-induced gut epithelial barrier dysfunction. In humans, several studies have demonstrated that vitamin D deficiency is correlated with increased severity of chronic liver disease. In humans, Vitamin D deficiency has been associated with increased liver damage and mortality in ALD.
Vitamin A
Consumption of alcohol decreases hepatic levels of vitamin A, with severity of deficiency correlating with the severity of liver disease. Vitamin A deficiency can lead to night blindness, hypogonadism, hypogeusia, and insomnia. Interestingly, vitamin A supplementation can be hepatotoxic with hepatotoxicity amplified by alcohol use. Thus, supplementation with vitamin A should be used with caution.
Magnesium
Magnesium deficiency is common in patients with chronic liver disease and occurs due to malabsorption of magnesium in the small intestine (one-third is absorbed in the small bowel) as well as inappropriately high renal excretion. Diuretic use further contributes to magnesium deficiency. Magnesium is a cofactor for over 200 enzymes and is required for many cellular processes as well as thiamine activation. Magnesium deficiency has the potential to increase oxidative stress by promoting iron accumulation and the production of perozinitrate.
In chronic liver disease, magnesium deficiency is associated with poor cognitive performance and reduced muscle strength. One study that included 13,504 patients evaluated the association between dietary magnesium intake and mortality, with findings that high intake of magnesium was associated with reduced risk of mortality due to liver disease. Another recent large cohort study exploring the association of magnesium intake and liver fibrosis found an inverse association between high total magnesium intake and significant fibrosis.
Zinc
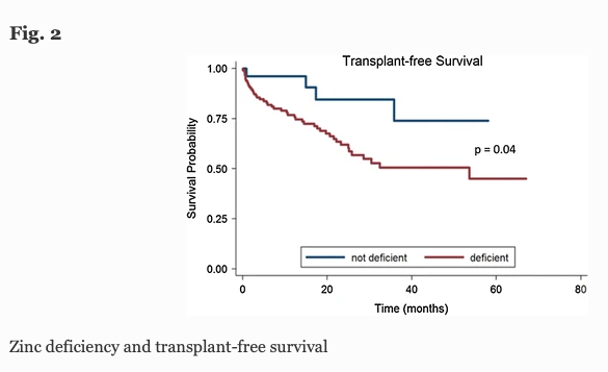
Zinc deficiency is found in >80% of patients with cirrhosis, with symptoms including muscle cramps, taste alteration, hepatic encephalopathy, and infections. The table below shows the correlation between serum zinc level and complications of cirrhosis in 163 patients, with lower levels of serum zinc significantly associated clinically with ascites, encephalopathy, and infection, as well as with diuretic and lactulose use.
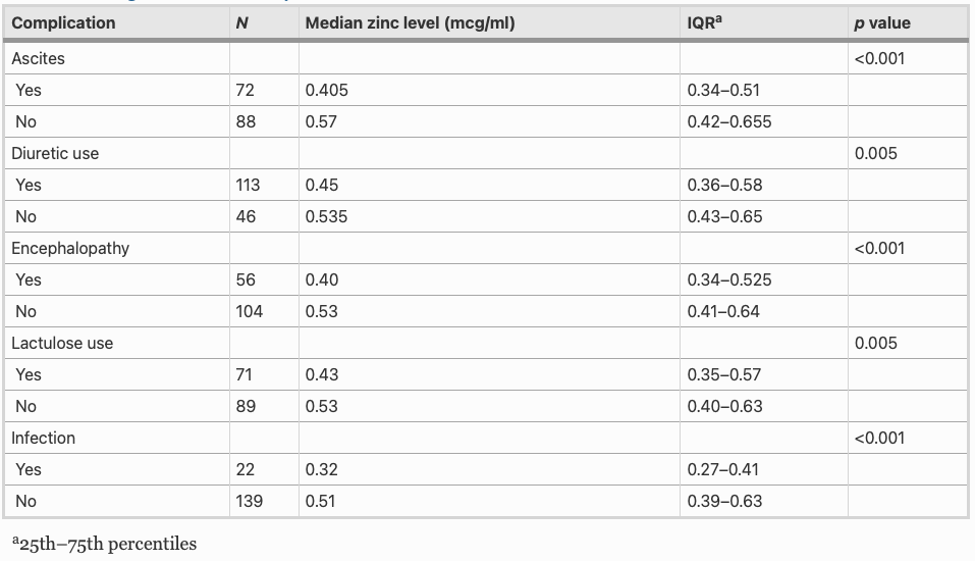
This study also found that patients with zinc deficiency had lower rates of 1- and 3-year transplant free survival, with 77% and 51% of deficient patients surviving at 1 and 3 years versus 96% and 74% of non-zinc deficient patients, respectively. This comparison is illustrated in the Kaplan-Meier curve below.
Zinc homeostasis is primarily regulated through absorption and excretion in the gastrointestinal track, with the liver playing a central role in homeostasis. Zinc is an essential trace element that acts as cofactor for alcohol dehydrogenase and is involved in cell signal transduction and gene expression. Zinc deficiency has been theorized to increase the amount of ethanol metabolized via CYP 2E1 and produce oxidative stress. Zinc deficiency has also been linked with reduced fatty acid beta-oxidation, impaired autophagy and hepatocyte apoptosis, lipid peroxidation, reduced tissue necrosis factor-alpha production, and increased circulating endotoxin. In animal models, zinc deficiency has been linked with increased fibrosis and steatosis with zinc supplementation improving steatosis, hepatocyte necrosis, and markers of fibrosis. In mice, zinc supplementation attenuated oxidative liver injury caused by ethanol. In humans, zinc deficiency has been associated with reduced levels of antioxidants as well as higher serum liver associated enzymes.
How does nutrition influence clinical outcomes?
Hepatic Encephalopathy and Other Clinical Outcomes
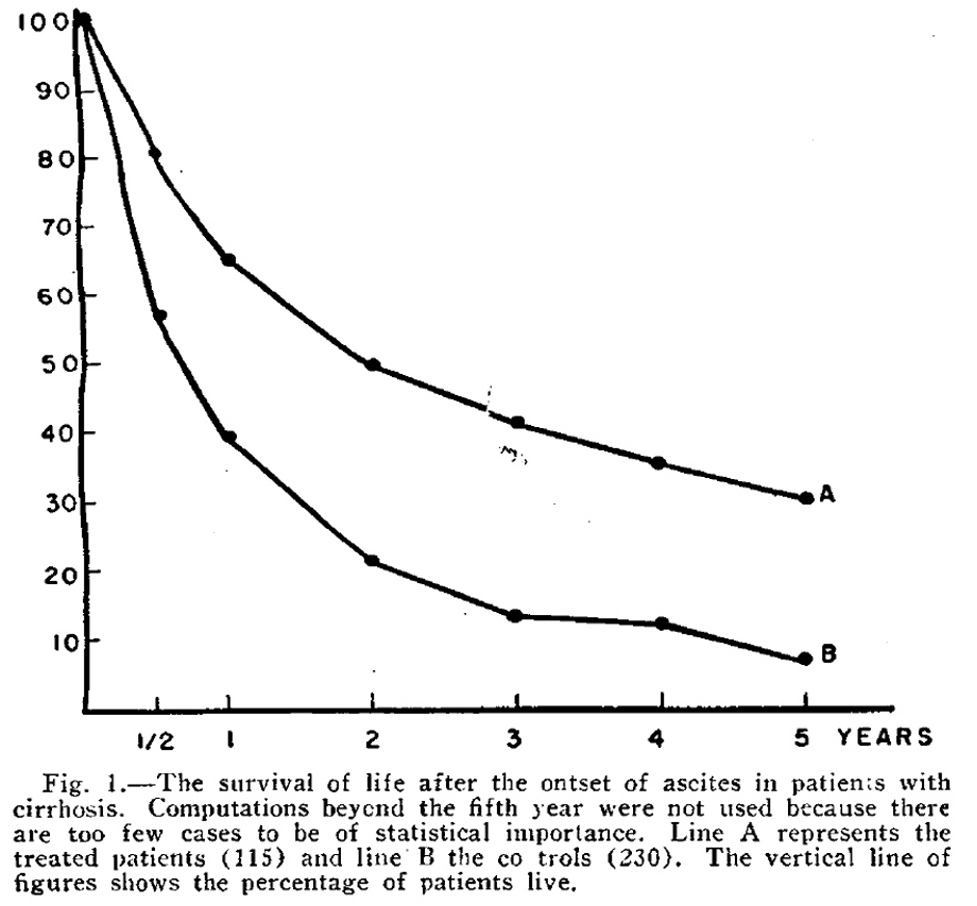
The composition of the ideal diet for patients with liver disease has been a subject of controversy since the 1930s. Patek et al demonstrated in 1948 that a high-protein (140 g) and vitamin B complex supplemented diet in patients with cirrhosis and ascites led to improved survival. In this study, 124 patients were treated with a high protein diet (Group A) and compared with 368 patients who were treated with the conventional high carbohydrate, low protein, low fat diet (Group B) and were followed for 10 years. The 5-year survival curve is shown below, depicting the percentage of patients surviving at various time points between the treatment group (A) and control group (B). Despite following patients for up to 10 years, this figure stopped at 5 years as many of the patients had died, with only 30% of treated patients alive at 5 years versus 7% of controls.
Despite the initial data supporting a high protein diet, there was a predominant belief that high protein diets precipitated hepatic encephalopathy. One 1954 case series of 3 patients with cirrhosis due to alcohol suggested that providing additional protein to these patients (increasing from 50g to 75 g daily) can induce or aggravate hepatic encephalopathy. The authors also suggested that withdrawal of protein may contribute to occasional remission. Low protein diets persisted in the treatment of liver disease well into the 1970s, with the widespread belief that that feeding protein to critically ill patients with liver disease, especially to those who may have impaired urea synthesis, created a positive nitrogen balance and aminoacidemia leading to hepatic encephalopathy.
This myth of dietary protein aggravating hepatic encephalopathy was finally debunked in a pivotal 2004 RCT by Córdoba et al. In this study, 30 patients with cirrhosis admitted for hepatic encephalopathy were randomized to receive either a low protein or normal protein diet for 14 days with no difference in hepatic encephalopathy between the two treatment groups.
Specific to alcohol use and hepatic encephalopathy, acetaldehyde, the immediate oxidative product of ethanol, impairs hepatic ureagenesis, which is the primary pathway for breakdown of ammonia. This in turn can lead to worsening hepatic encephalopathy.
Sarcopenia
Sarcopenia is the loss of skeletal muscle mass and occurs in up to 60% of patients with chronic liver disease. The European Working Group on Sarcopenia defines the disorder as “a progressive and generalized skeletal muscle disorder associated with an increased likelihood of adverse outcomes including falls, fractures, disability, and mortality.” Malnutrition is one of many factors that contributes to the development of sarcopenia.
Skeletal muscle mass is maintained by a balance of protein synthesis and proteolysis. As we know, cirrhosis is a catabolic state that is characterized by an “accelerated starvation,” with early gluconeogenesis and accelerated consumption of amino acids as a source of energy. This accelerated skeletal muscle protein loss and reduced muscle protein synthesis results in sarcopenia.
Sarcopenia is consistently identified as a predictor of both pre-and post- transplant mortality risk, with the presence of sarcopenia associated with poor clinical outcomes such as decompensation, hepatic encephalopathy, sepsis, length of hospital stay and prolonged post-transplant ICU admission, as well as increased healthcare costs. Patients with ALD have higher prevalence of sarcopenia than other etiologies of chronic liver disease, with sarcopenia affecting up to 80% of patients with decompensated cirrhosis due to alcohol compared to 60% of patients with decompensated cirrhosis due to other etiologies. This observation is likely due to the combination of effects that alcohol and its metabolites have on skeletal muscle. While the majority of ethanol is metabolized in the liver, some ethanol metabolism occurs in muscle tissue, leading to reactive oxygen species and mitochondrial dysfunction. Acetaldehyde, an alcohol metabolite, impairs skeletal muscle synthesis and increases autophagy.
How should we address malnutrition?
The European Societies for Clinical Nutrition and Metabolism guideline on clinical nutrition and liver disease, Malnutrition, Frailty, and Sarcopenia in Patients With Cirrhosis: 2021 Practice Guidance by the American Association for the Study of Liver Diseases, and the EASL Clinical Practice Guidelines on nutrition in chronic liver disease recommend that all patients with liver disease be screened or malnutrition using a validated tool, such as the Royal Free Hospital Nutrition Prioritizing Tool.
Patients with ALD and malnutrition should be encouraged to increase oral intake, with a goal 45-40 kcal/kg body weight and protein intake of 1.5 g/kg body weight, with body weight ideally representative of the patient’s dry body weight. Patients who continue to drink alcohol should be encouraged to abstain from alcohol to both prevent ongoing liver injury as well as to improve nutritional status. Given the reduced glycogen storage and accelerated gluconeogenesis, patients with alcohol-associated liver disease need to be encouraged to avoid fasting with frequent, small meals and prioritization of early breakfast and a bedtime snack.
The late evening snack has been studied extensively in chronic liver disease. Given that the longest period of physiologic fasting is between dinner and breakfast, inserting a late evening snack extends the postprandial state and preserves muscle mass by reducing the utilization of amino acids from skeletal muscle in gluconeogenesis as well as increasing the rate of skeletal muscle protein synthesis. A 2008 randomized control trial that included 103 outpatients patients with cirrhosis of varying etiologies (including 15 patients with ALD) demonstrated that long term administration of a late-night snack increased total body protein and increased lean body mass when compared to a daytime snack. The following figure shows change in total body protein at 3, 6, and 12 months in patients who were given daytime versus nighttime snacks.
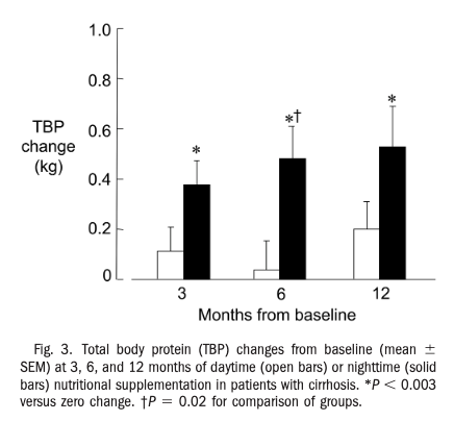
Interestingly, patients who were randomized to receive evening snacks also had improved health related quality of life scores when compared to patients who were given day-time snacks and experienced these changes earlier on in treatment course.
In patients not able to maintain nutrition with oral intake, supplemental nutrition with either nasogastric tube or parenteral nutrition may be warranted, with enteral nutrition preferred. In a 2016 RCT investigating intensive enteral nutrition in patients with severe AAH treated with corticosteroids, Moreno et al found no significant difference in 6-month mortality between patients treated with intensive enteral feeding and methylprednisolone compared to patients who just received methylprednisolone. However, low daily calorie intake was associated with higher morality and, conversely, improved 6-month survival was associated with ≥65 g/d of lipids and ≥77.6 g/d of protein intake. On closer look, there was a non-significant lower mortality rate at 1 and 6 months in the intensive nutrition group compared to the control. Interestingly, enteral feeding was difficult to implement, with nearly half of patient in this group withdrawing from the feeding tube prematurely, which may have contributed to the negative trial results.
A 2012 Cochrane Review on nutritional support for liver disease that included parenteral nutrition in ALD, found that parenteral nutrition decreased total serum bilirubin, improved nitrogen balance, but has not been shown to improve mortality or hepatic encephalopathy.
How are we doing?
Unfortunately, despite these recommendations, the opportunity to intervene nutritionally on these patients is often missed. In a cohort study of 231 hospitalized patients with cirrhosis, only 57% had a nutritional assessment during their admission. Of these patients assessed by a dietitian and determined to be malnourished, only 38% received supplementation with either oral nutritional supplements, enteral, or parenteral nutrition. The remaining 62% of patients received dietary advise alone with no concrete intervention. In another study that included 53 inpatients hospitalized with AAH, only 55% had nutrition evaluations and only 17% had appropriate diet orders.
Key Points
- Malnutrition and micronutrient deficiencies are very common in patients with ALD and are associated with poor outcomes
- Evaluate for malnutrition with the help of a registered dietitian
- If a patient is malnourished, intervene!
- Remember that patients with cirrhosis benefit from high protein high calorie diet as well as a late-night snack
- Interventions should be personalized to patient and individual needs