Pathology Pearls: Alpha-1 antitrypsin deficiency

Brief Case Presentation
A 50-year-old male with a 30 pack-year smoking history and chronic obstructive pulmonary disease (COPD) status-post lung transplant presents for evaluation of mild hypertransaminasemia on routine follow-up. His most recent lung biopsy showed no features of acute cellular rejection. Current medications include tacrolimus and mycophenolate. A pre-biopsy ultrasound of the liver showed mild heterogeneity and coarsening of the hepatic echotexture, suggestive of underlying hepatic fibrosis.
Selected lab values are as follows:
| Lab Value | Result | Reference Range |
| Hgb | 14.7 g/dL | 13.0 - 17.5 g/dl |
| WBC | 9.4 K/uL | 3.5 - 10.5 K/uL |
| Platelets | 144 K/uL | 140-390 K/uL |
| ALT | 79 U/L | 0 - 52 U/L |
| AST | 44 U/L | 0 - 39 U/L |
| Alkaline phosphatase | 110 U/L | 34 - 104 U/L |
| Albumin | 4.5 g/dL | 3.5 - 5.7 g/dL |
| Total bilirubin | 0.6 mg/dL | 0.0 - 1.0 mg/dL |
| aPTT | 29.2 s | 25 - 35 s |
| PT / INR | 12.9s 1.1 |
10.5 - 13.5 s 0.8 - 1.2 |
| HAV | Negative | - |
| Total HBV core Ab | Negative | - |
| HBV surface Ab | Reactive | - |
| HBV surface Ag | Negative | |
| α1-antitripsin level, serum | 74 mg/dL | 83 - 199 mg/dL |
Liver Biopsy Findings
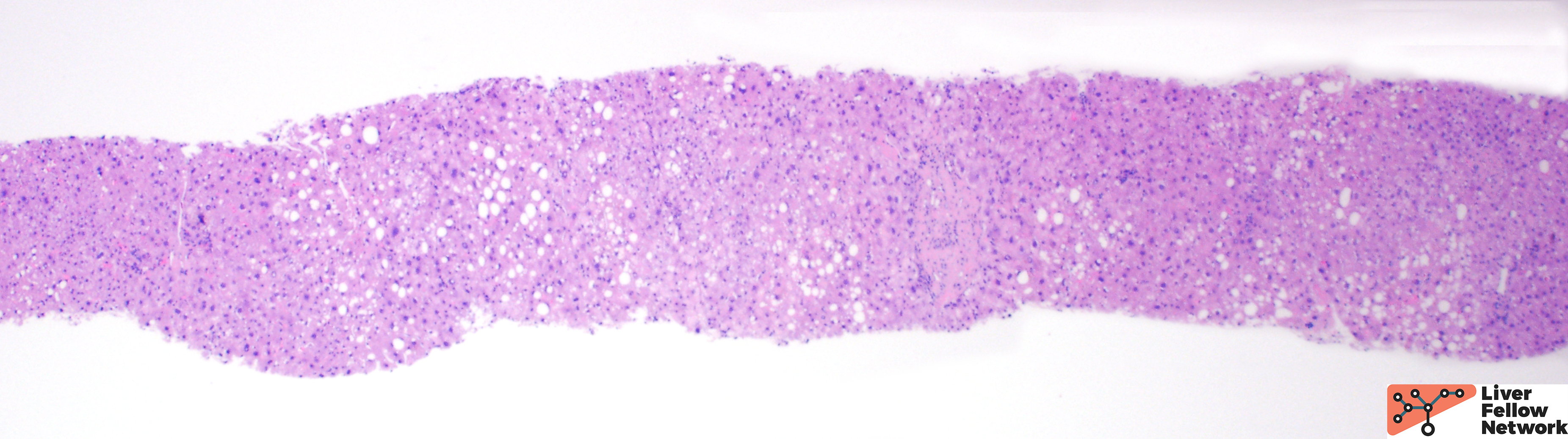
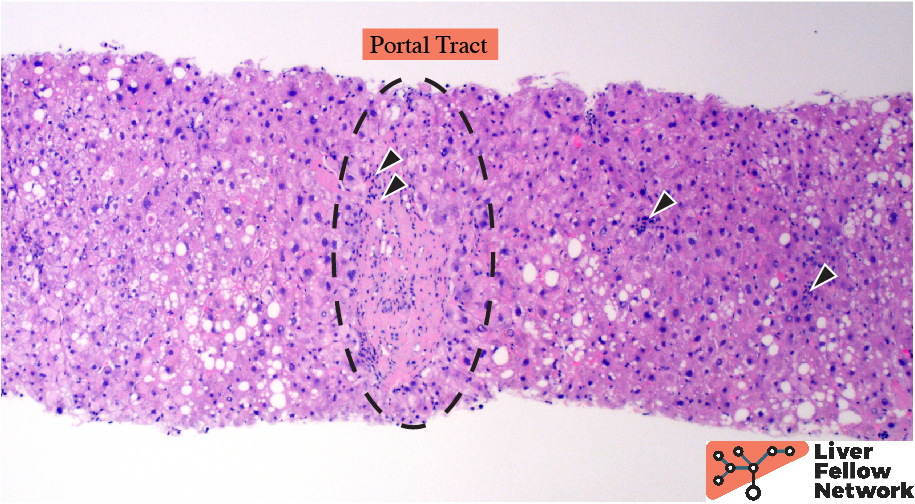
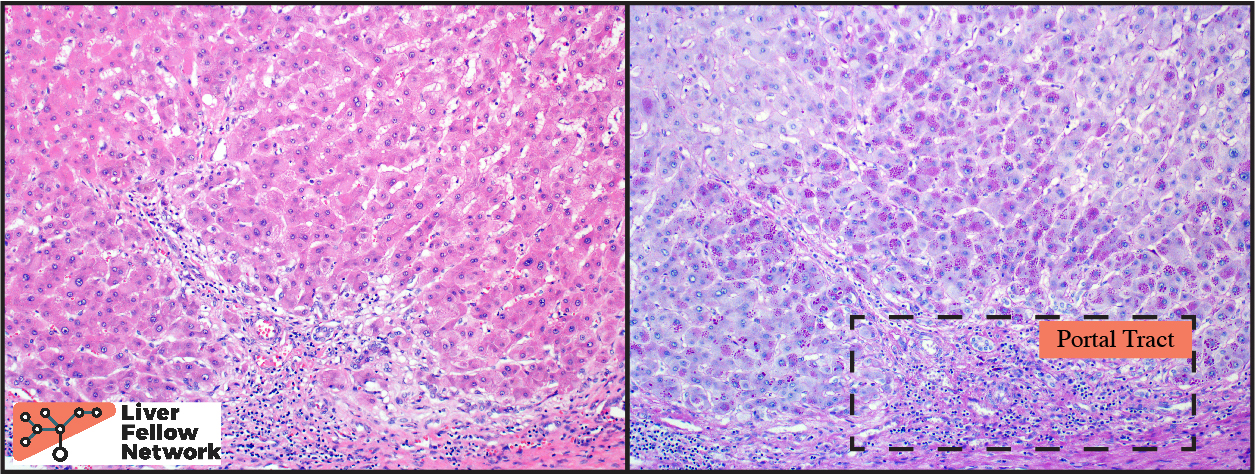
At low power, the liver core shows very mild portal and lobular lymphocytic infiltrate and mild macrovesicular steatosis of the hepatocytes (Figures 1 and 2).
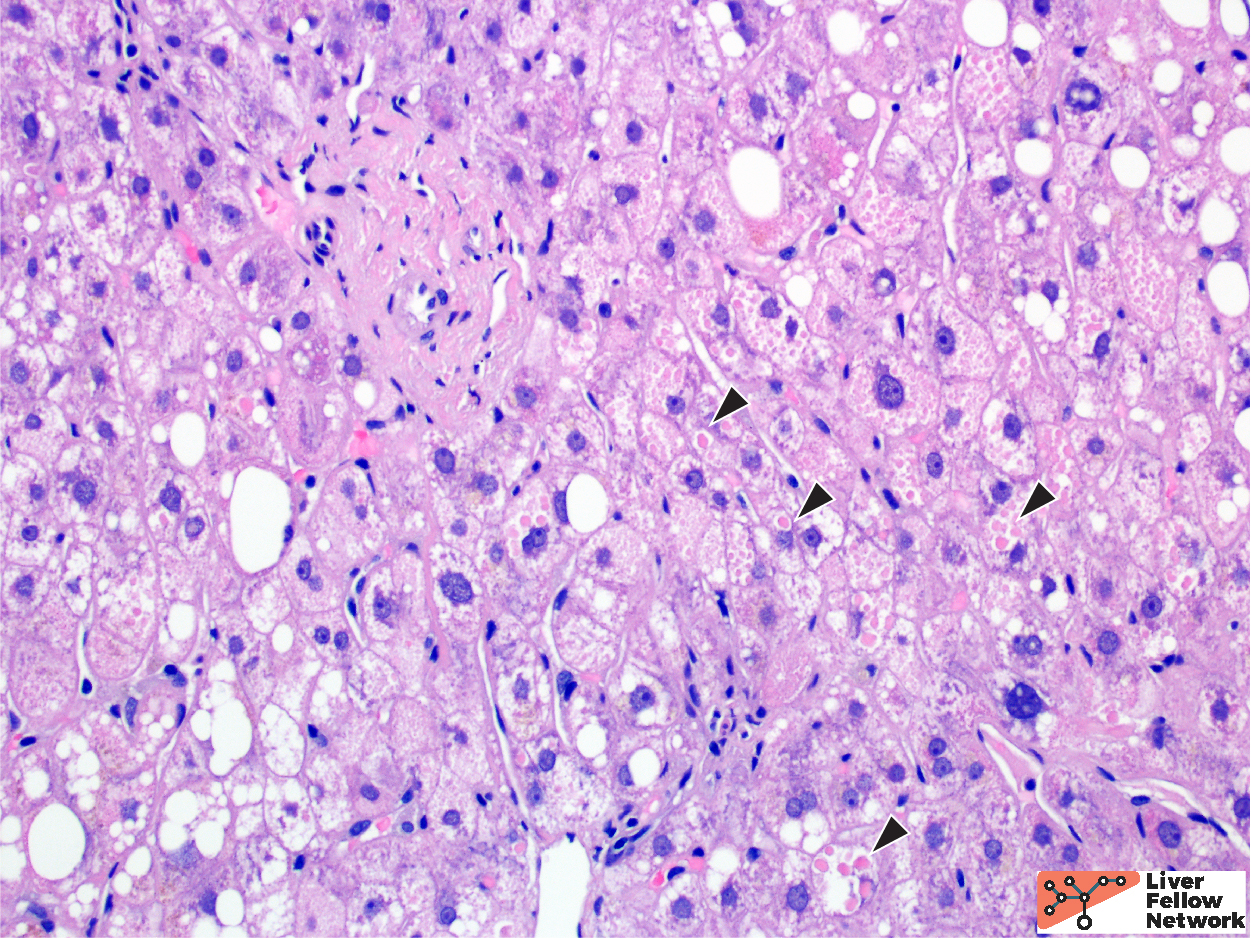
At medium to high power, hepatocellular cytoplasmic inclusions are identified, comprised of round eosinophilic globules (Figure 3).
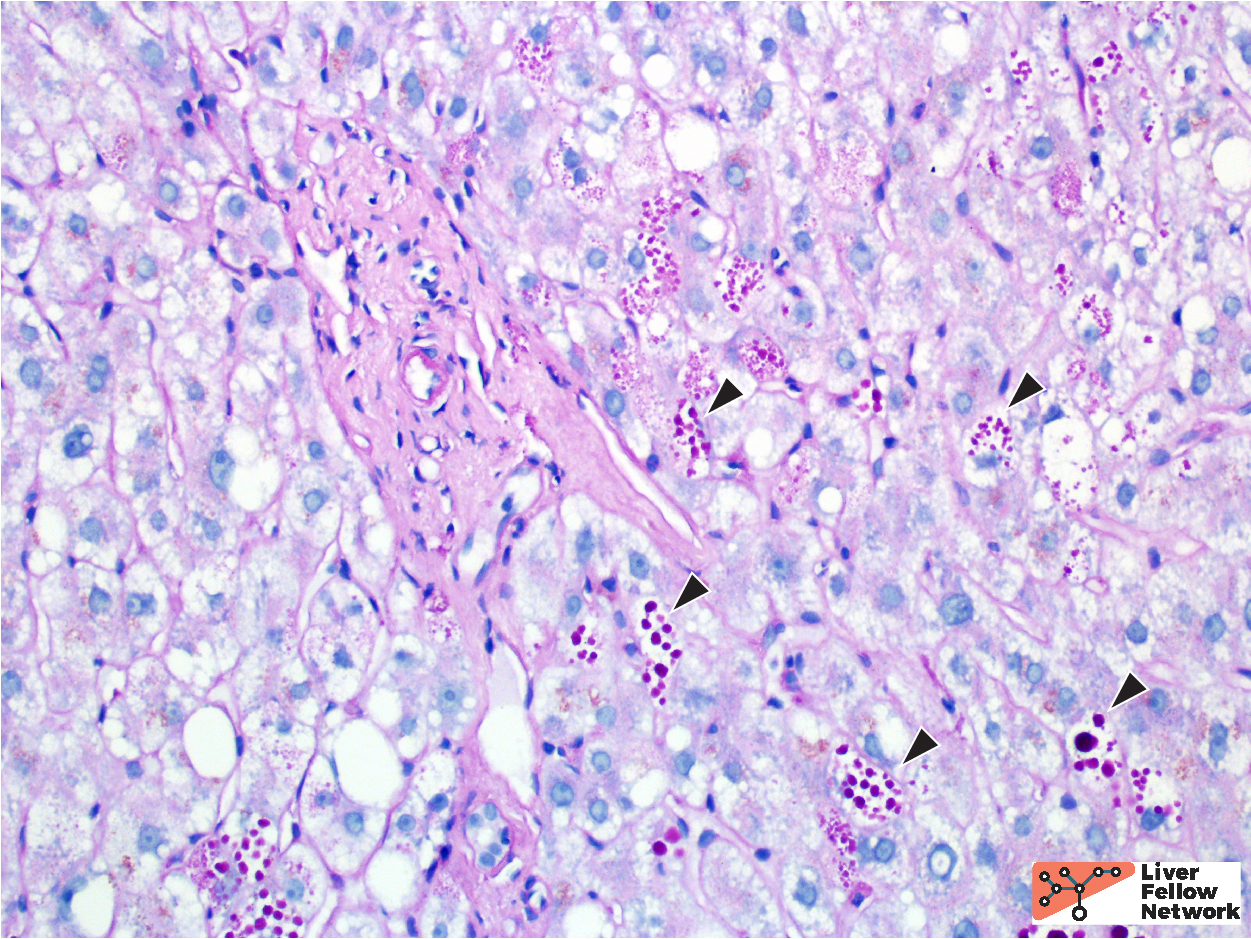
A PAS stain with diastase (PAS/D) highlights the hepatocellular inclusions and helps demonstrate a peri-portal (zone 1) distribution of the globules (Figure 4). An immunohistochemical stain against the α1-antitrypsin protein demonstrates that the globules are comprised of aggregates of α1-antitrypsin protein product (Figure 5).
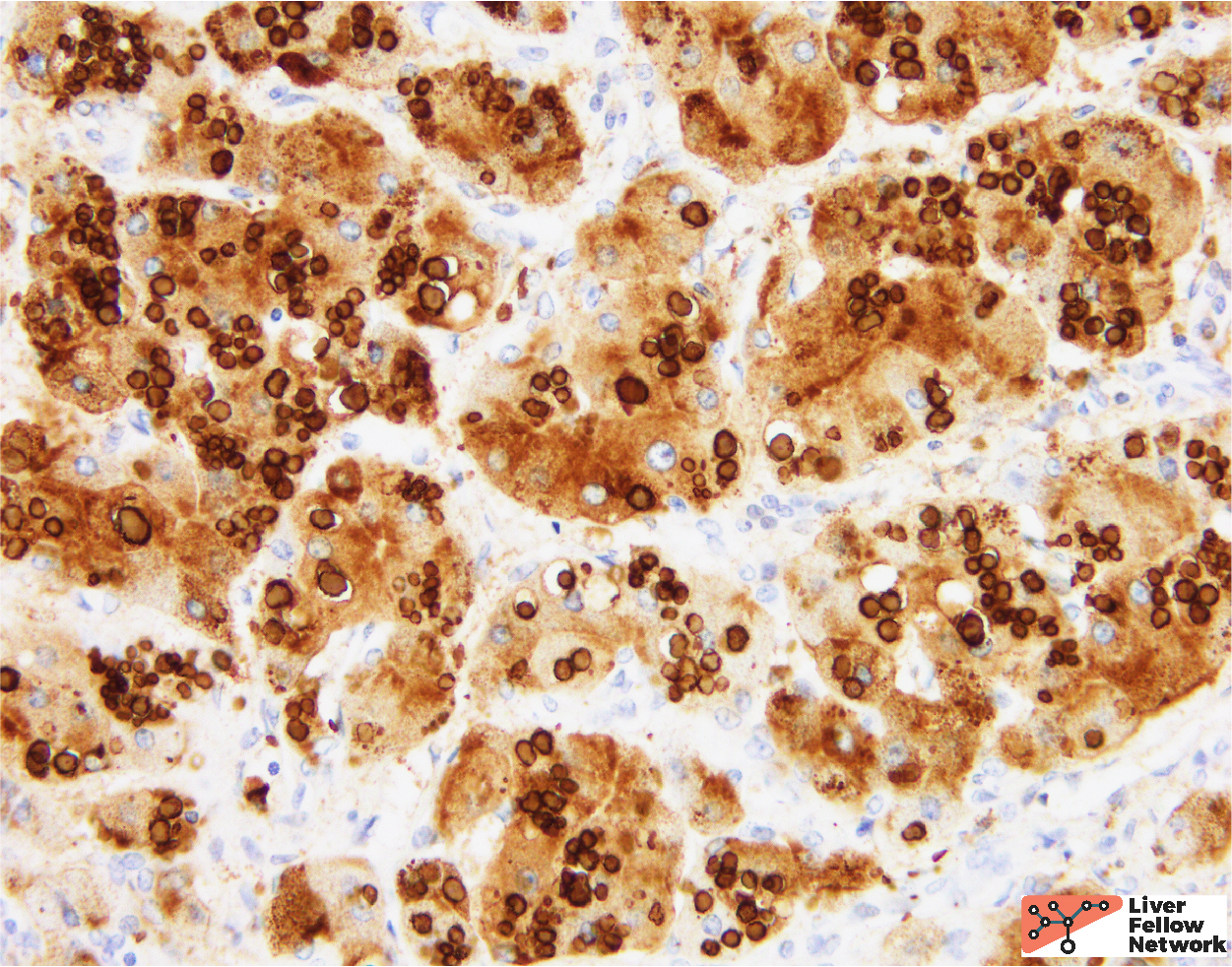
In the clinical context provided, the histologic findings are compatible with α1-antitrypsin deficiency.
Etiology and Molecular Characteristic of α1-antitrypsin deficiency
α1-antitrypsin deficiency (A1AT) is an inherited metabolic disorder characterized by mutations in the SERPINA1 gene on chromosome 14 that encodes the protease inhibitor (PI) α1-antitrypsin. Mutations result in an abnormal protein conformation leading to aggregation within hepatocytes (the primary site of A1AT expression), and decreased levels of circulating A1AT. The major target of A1AT is leukocyte elastase, and increased functional elastase causes clinical symptoms, including pulmonary emphysema and variable liver disease, ranging from clinically silent to severe cirrhosis.
The wild-type allele of SERPINA1 is referred to as the M allele. The two most common mutations occur at residue 342 (Gly->Lys) and residue 264 (Glu-> Val), and are referred to as the Z allele and S allele, respectively. Phenotypically, A1AT is measured by the degree of reduction in A1AT. The Pi*MM phenotype is the wild-type phenotype, with normal serum A1AT levels. The most prevalent homozygous state is Pi*ZZ, associated with severely decreased A1AT serum levels; Pi*ZZ is identified in >90% of patient with clinically relevant disease. Heterozygous states (Pi*MZ, Pi*MS, or Pi*SZ) are associated with modest reduction in serum A1AT and are often clinically silent.
Confirmation of the diagnosis requires molecular testing via polymerase chain reaction (PCR) mutation-specific amplification or DNA sequencing (i.e., via next generation sequencing, NGS) to confirm the genotype of the individual.
Histologic Features of α1-antitrypsin deficiency
While the histologic features of α1-antitrypsin (A1AT) deficiency in adults is variable, the most characteristic feature is abnormal hepatocellular cytoplasmic eosinophilic globules. These represent misfolded A1AT protein, which aggregates in the endoplasmic reticulum.
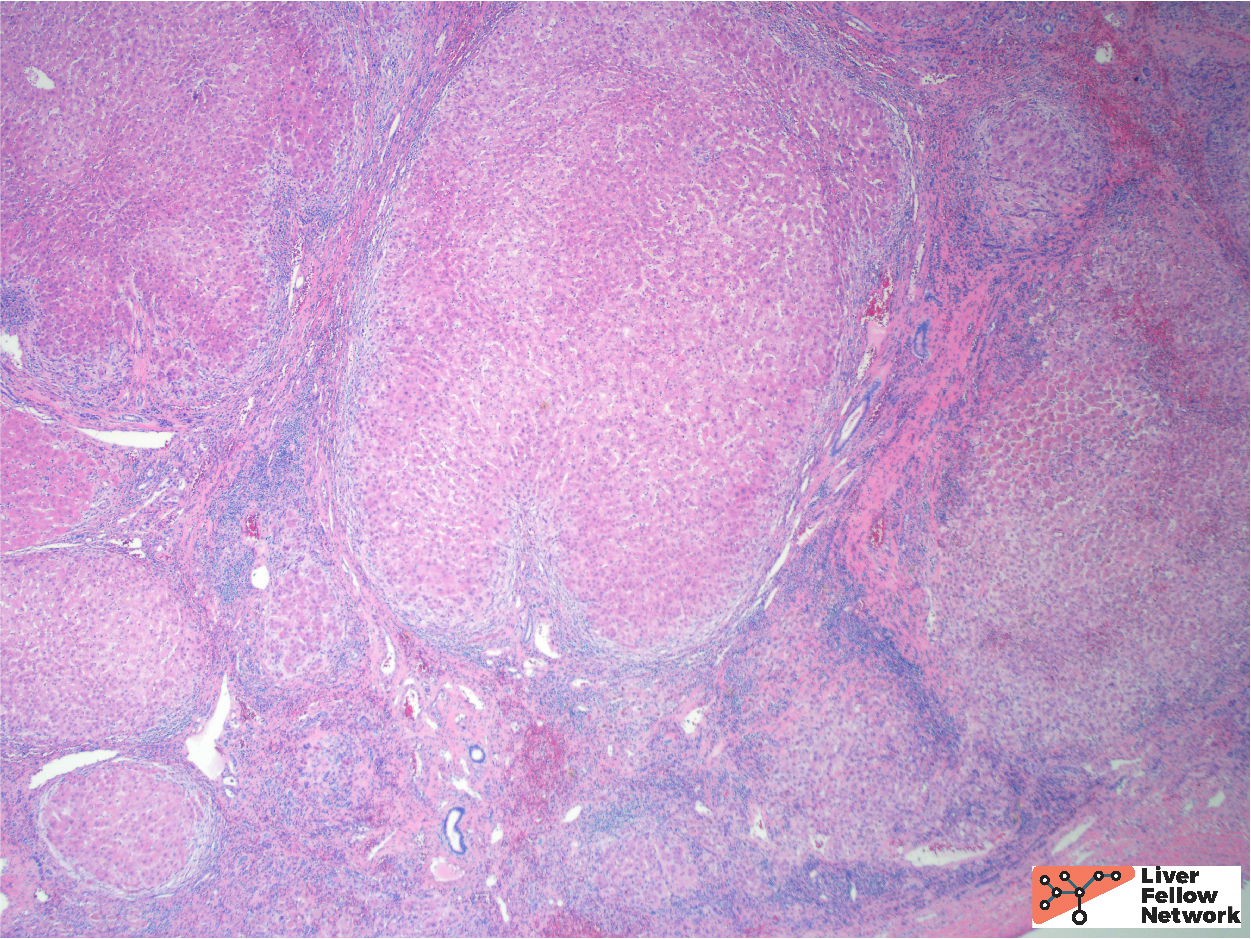
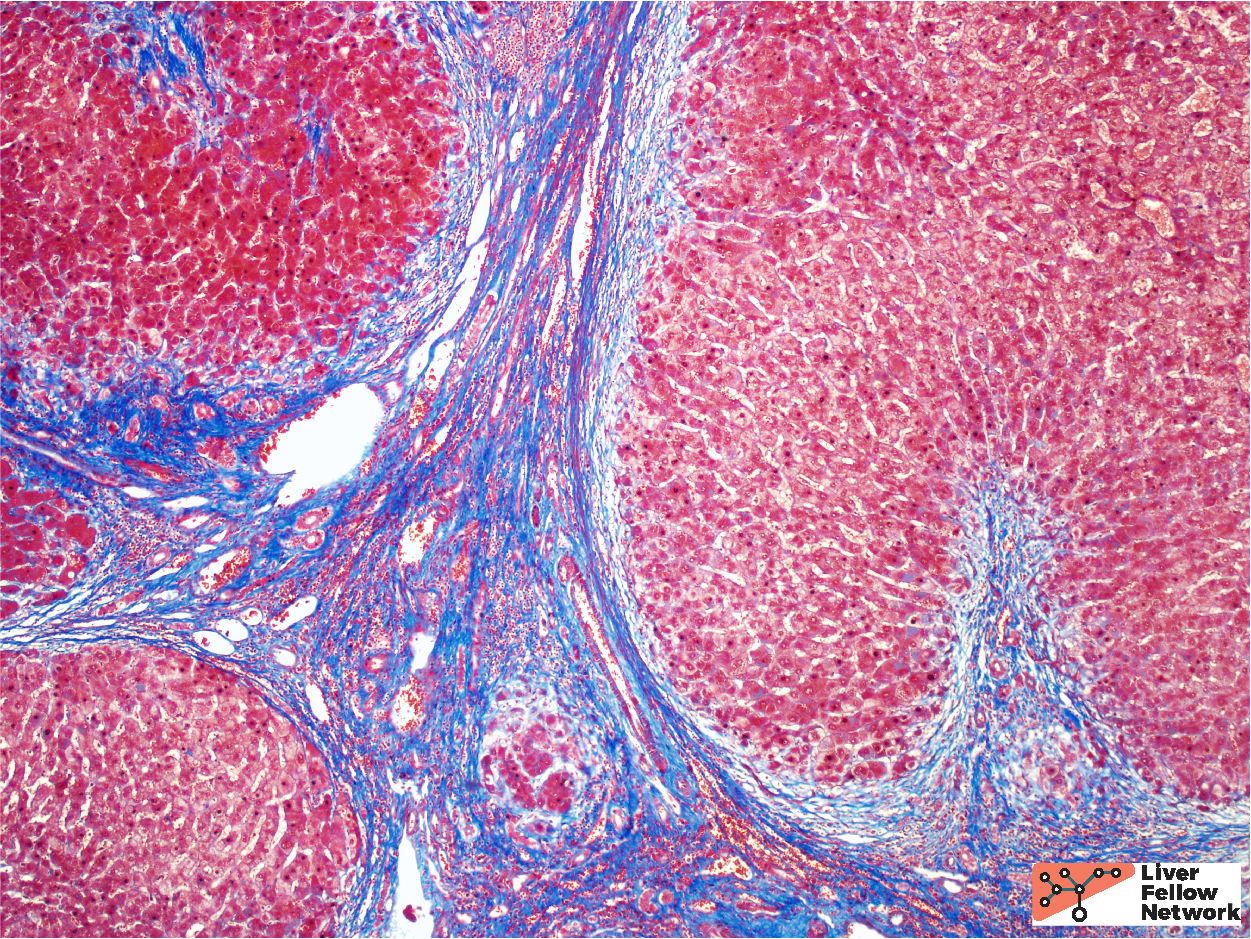
The background features of A1AT deficiency include variable portal lymphocytic infiltration, variable hepatocellular steatosis, and variable cholestasis. Patients can progress to fibrosis and cirrhosis (~15% of PiZ individuals) and the risk of cirrhosis increases with age. Patient factors, such as obesity and alcohol consumption, can increase the risk of progression to cirrhosis. Figures 6 through 8 illustrate another case of A1AT deficiency in a liver explant. In this instance, the effects of A1AT deficiency in a heterozygous individual was compounded by non-alcoholic steatohepatitis (NASH); the liver progressed to severe fibrosis and cirrhosis eventually requiring liver transplantation.
Differential Diagnosis for α1-Antitrypsin Deficiency
Because eosinophilic round globules are not pathognomonic for a1-antitrypsin deficiency, the top differential diagnostic considerations include conditions that produce globules that mimic A1AT globules. Other sources of cytoplasmic globules include congestion associated globules (associated with centrilobular distribution, rather than periportal distribution seen in A1AT), aggregates of mega-mitochondria (PAS/D negative), lipofuscin pigment (red-brown color on PASD stain), and immunoglobulin aggregates (associated with autoimmune hepatitis and primary biliary cirrhosis, will be highlighted by immunostaining for immunoglobulin). Lafora disease is a rare source of pale, kidney-shaped inclusions that are PASD negative and positive on colloidal iron and silver stains.
References
- 1. Torbenson, Surgical Pathology of the Liver. 2018.
- 2. Odze and Goldblum, Surgical Pathology of the GI Tract, Liver, Biliary Tract and Pancreas: Third Edition. 2014
- 3. Nussbaum, Thompson and Thompson Genetics in Medicine: Eighth Edition. 2015.
- 4. Burt, MacSween's Pathology of the Liver: Seventh Edition. 2017.