Pathology Pearls: Autosomal dominant polycystic kidney disease/ Polycystic Liver Disease

Brief Case Presentation
The patient is a 50-year-old African American with end-stage renal disease status post bilateral nephrectomies due to recurrent infection and on hemodialysis. She presented with early satiety, increasing abdominal fullness with pain, and unintentional weight loss. Family history includes hypertension and cardiac disease. Physical examination shows abdominal distention with tenderness. Her liver function tests and abdominal CT scan are shown below, and given the imaging findings, she was admitted for a planned left partial hepatectomy.
Lab
| Lab | Result | (Reference Range) | ||
| ALT | 32 | (6-45 IU/L) | ||
| AST | 80 | (10-42 IU/L) | ||
| Alkaline Phosphatase | 49 | (34-104 IU/L) | ||
| Total Bilirubin | 0.4 | (0.2-1.3 mg/dl) | ||
| Direct Bilirubin | 0.1 | (0.0-0.3 mg/dl) |
Abdominal CT
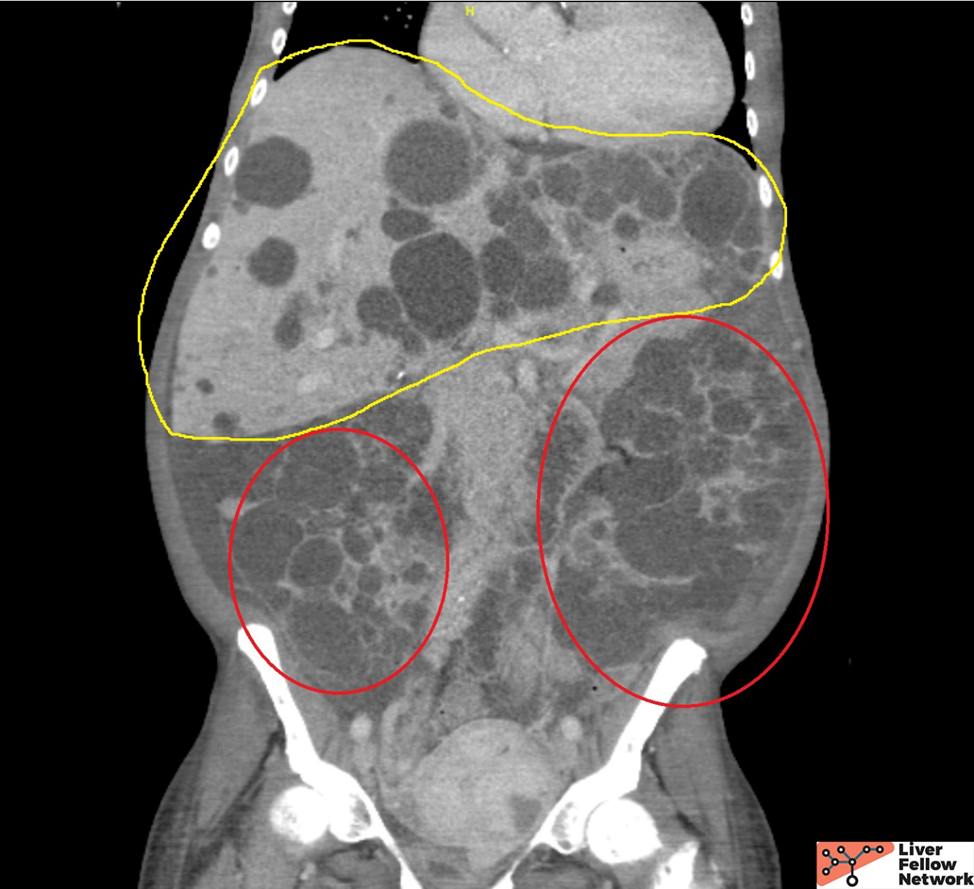
Hepatobiliary: Innumerable hepatic cysts. (Yellow outline)
Partial Hepatectomy Findings
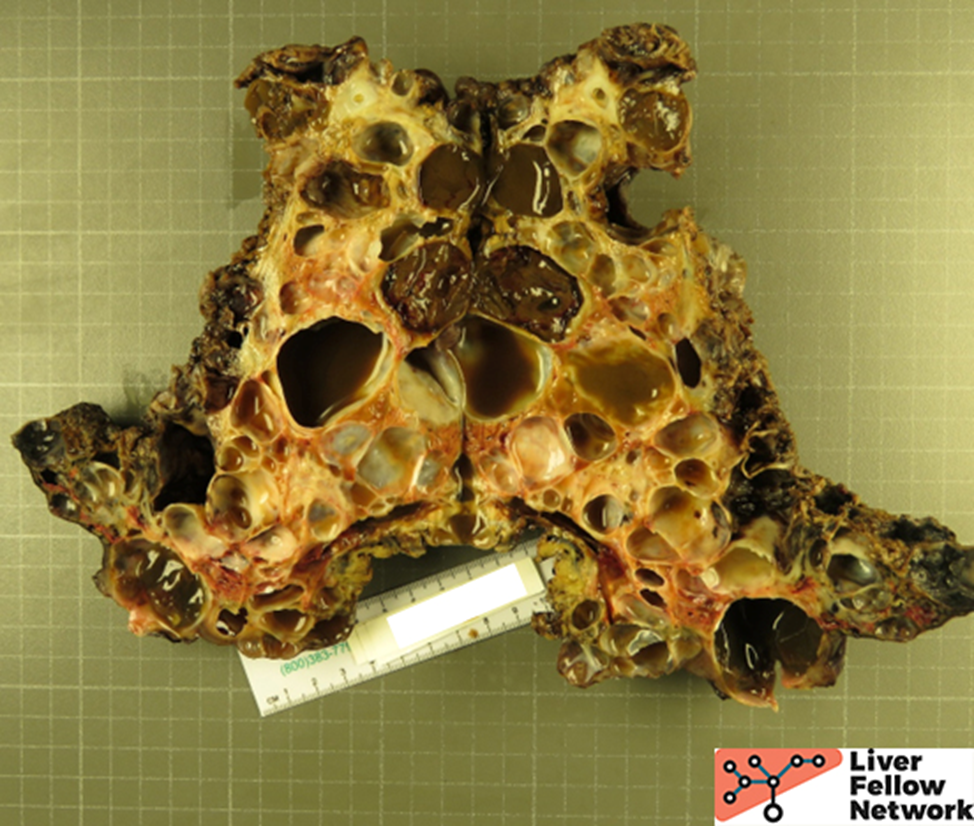
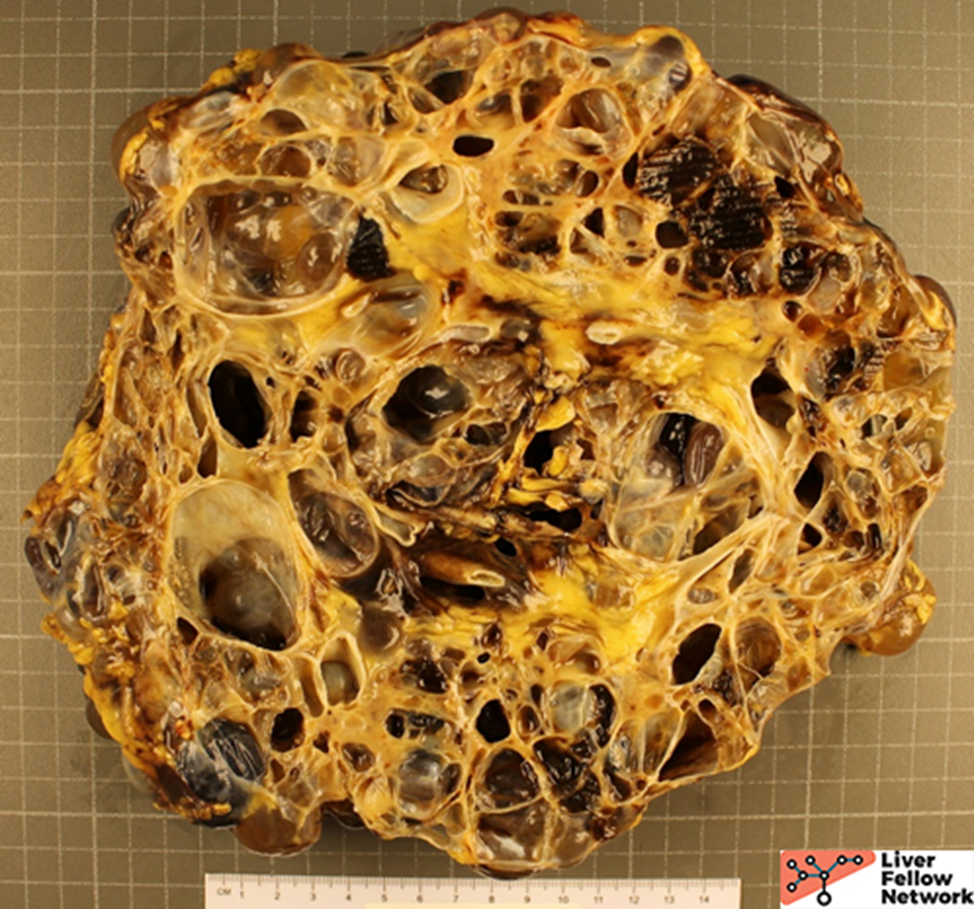
Gross Picture
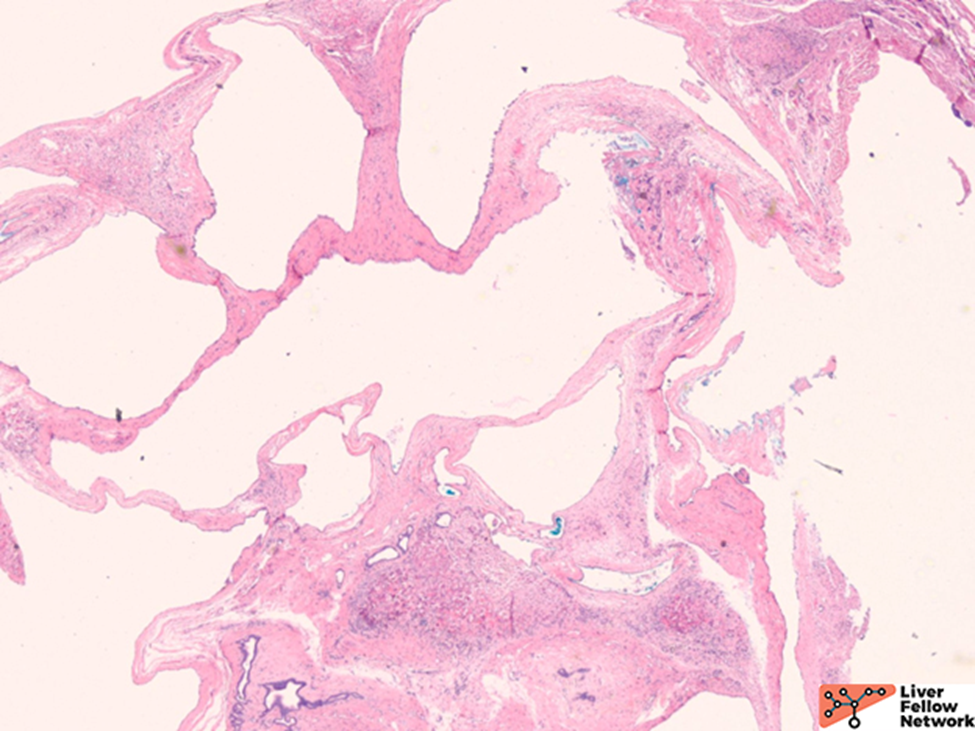
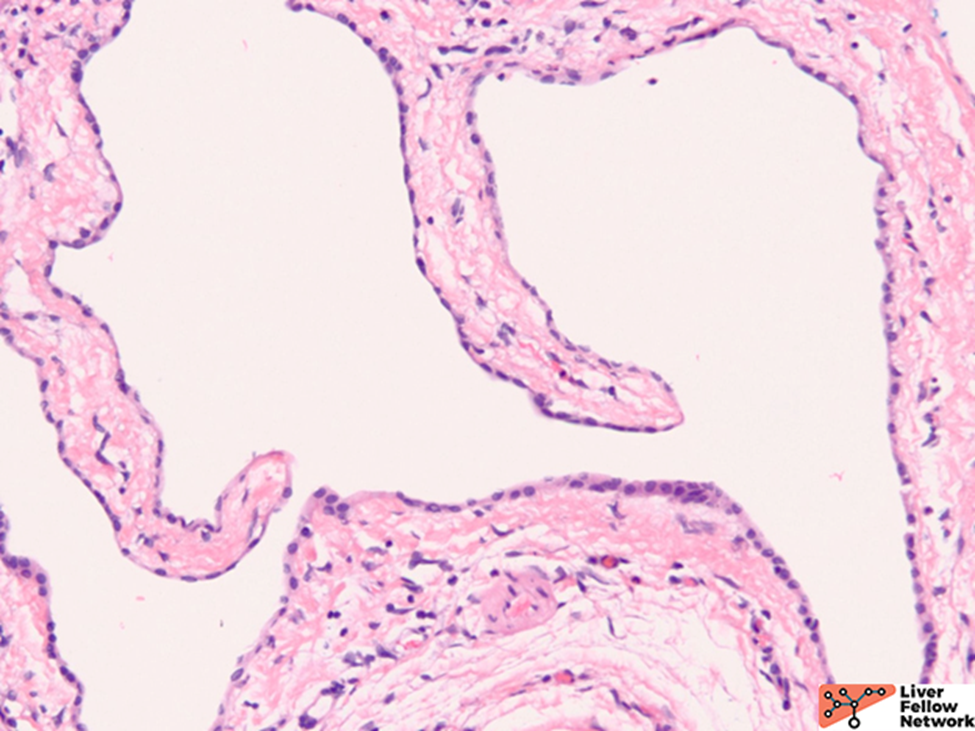
Microscopic Findings
Total nephrectomy findings
Gross Picture
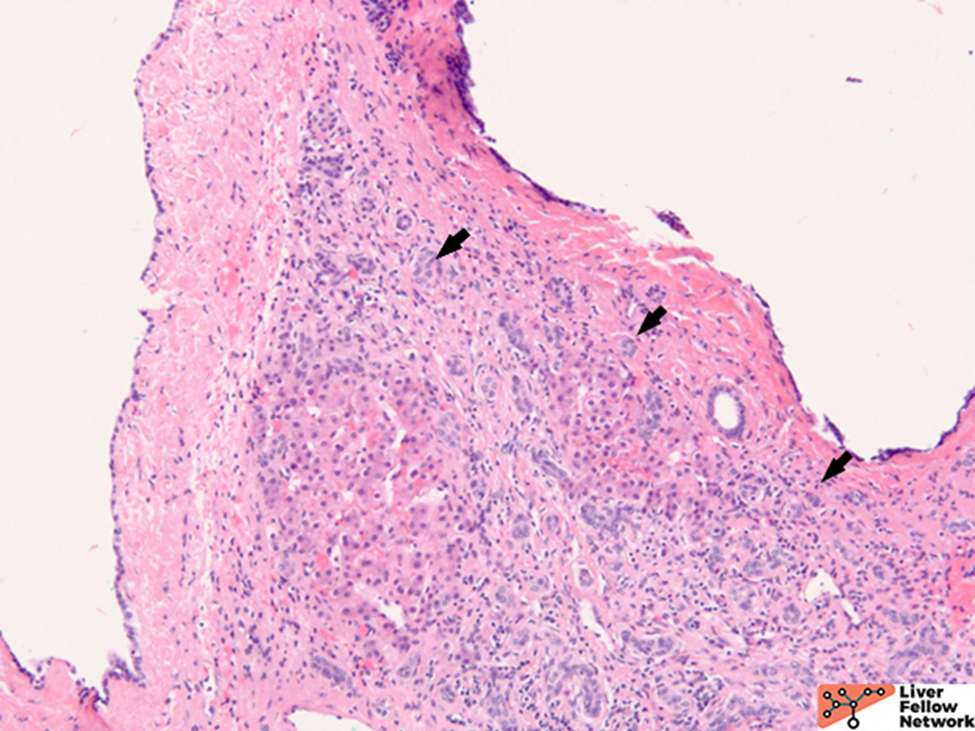
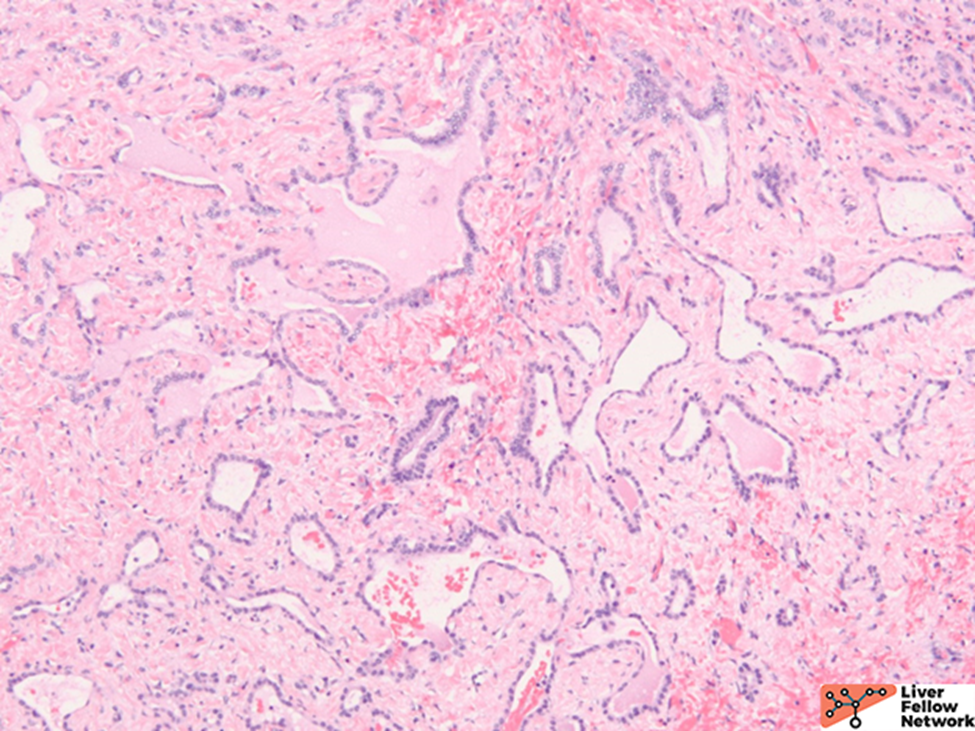
Microscopic Findings
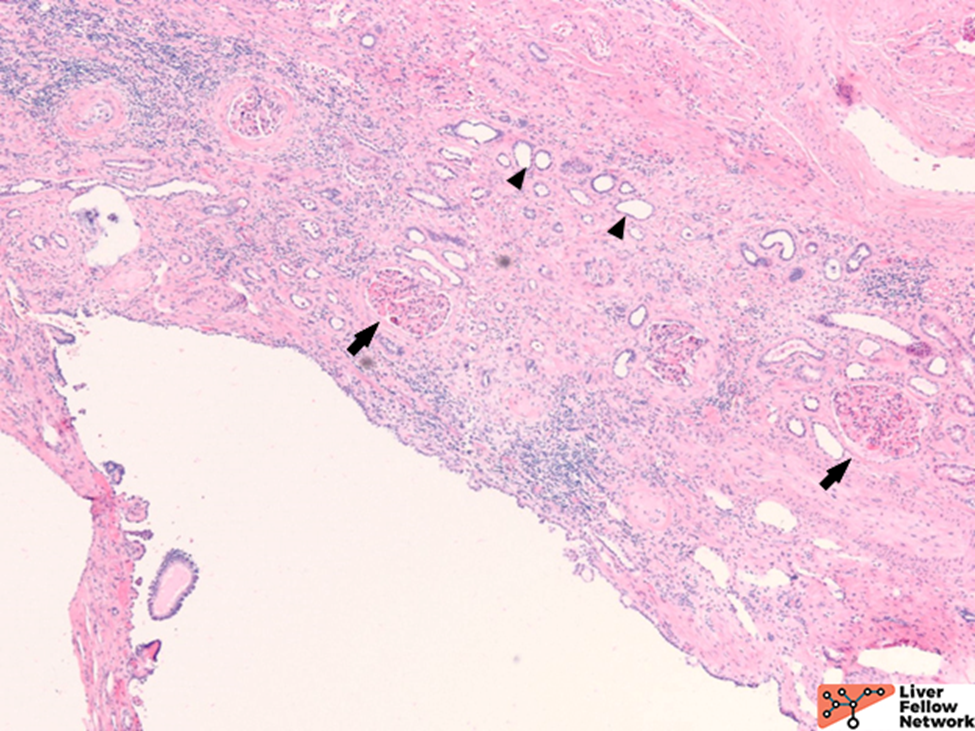
Although at the time of resection, there was no reported family history of adult polycystic kidney disease, the bilateral polycystic kidneys and liver cysts without other specific cystic disorders are sufficient to render the diagnosis of Polycystic Liver Disease as part of a component of Autosomal Dominant Polycystic Kidney Disease. However, genetic counseling is still essential. Further exploration through this patient’s family history revealed that her father and son had adult polycystic kidney disease as well.
Autosomal dominant polycystic kidney disease (ADPKD)/Polycystic Liver Disease (PLD)
Adult liver cystic lesions can be classified as hereditary, neoplastic, inflammatory, or mixed. The hereditary form of polycystic liver disease (PLD) is mostly associated with autosomal dominant polycystic kidney disease (ADPKD). It is also related to autosomal recessive polycystic kidney disease (ARPKD). Isolated PLD without renal cysts is rare and possesses distinct genetic features.1
ADPKD is the most common inherited renal disease, affecting 1 in 1000 live births and accounting for 8-10 percent of end-stage renal disease. ADPKD is a systemic disease with involvement of the kidneys, liver, cardiovascular system, and gastrointestinal system. Presenting manifestations include cerebral aneurysms, colonic diverticula, cardiac valve issues, and cysts within the liver, ovary, pancreas, spleen, and central nervous system.2 While some cases of PLD are discovered incidentally, many patients are identified due to family history of ADPKD.
The majority of ADPKD cases are secondary to a mutation in the PKD1 gene, which encodes the polycystin1 protein; a smaller number are related to mutations in the PKD2 gene encoding polycystin2.3
The exact mechanism of the cystogenesis is unclear, but it is believed that polycystin1 and 2 are related to primary cilia and function to inhibit cystogenesis.3
Clinical presentation of ADPKD includes hypertension, hematuria, proteinuria, kidney function impairment, and mass effect. In patients with a family history of ADPKD, imaging alone is sufficient for diagnosis. In patients without a family history, 10 or more cysts (≥5 mm) in each kidney or liver cysts without obvious features of a different cystic disorder are highly suspicious for ADPKD. Genetic testing by sequencing would be ideal to confirm the diagnosis.4 In ADPKD, most commonly hepatic cysts develop later than renal cysts. They are clinically insignificant, often presented for the first time in the fourth decade of life, and gradually increased in number and size. The clinical manifestations are predominantly determined by the number, size, location, and distribution of the liver cysts, which are related to the extent of hepatomegaly. The symptom includes pain, heartburn, early satiety, weight loss, and anorexia. Females with multiple pregnancies and prolonged estrogen exposure have a higher risk of liver symptoms. Other complications include hemorrhage, infection, portal hypertension due to altered structures, jaundice, and liver failure.5 Therapeutic interventions are not warranted in asymptomatic patients. Conservative treatment is recommended for most patients. For symptomatic patients, surgical intervention may be beneficial.6
Gross and histology of polycystic liver disease associated with ADPKD
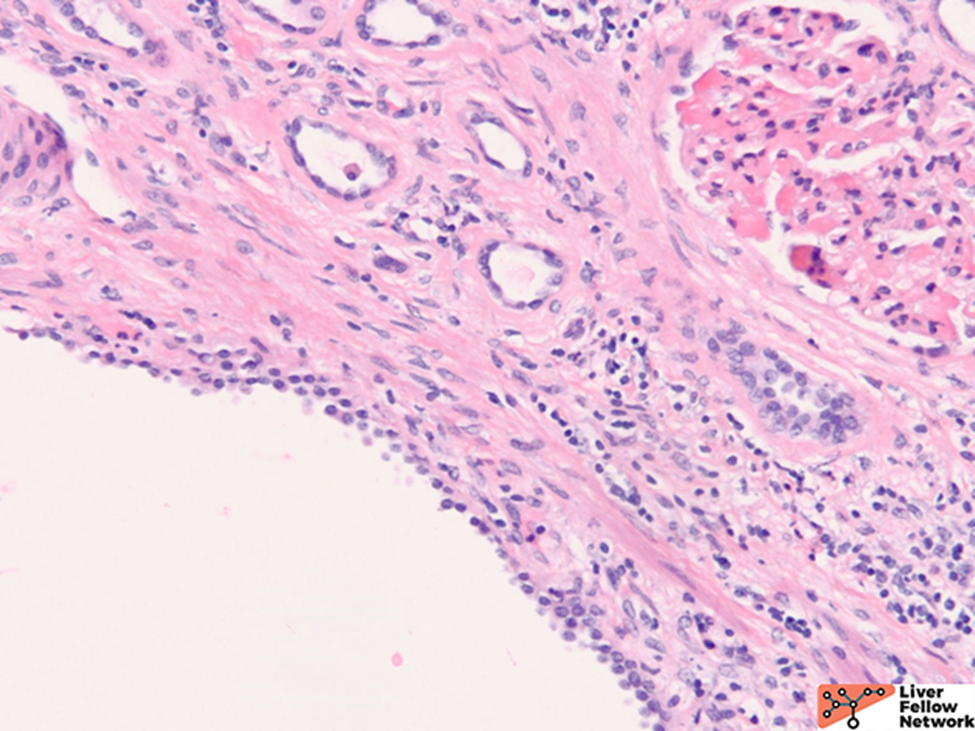
Grossly, there are multiple cysts in variable sizes filled with clear to yellow fluid replacing most of the hepatic parenchyma. Sectioning through the liver demonstrates the cysts lined by a smooth lining without papillary excrescences or masses.
Histologically, there are cystic spaces of various sizes occasionally accompanied with Von Meyenberg complexes, which showmis-shaped dilated bile ducts lined by cuboidal cells in a fibrotic stroma.
The lining of the cysts consists of simple cuboidal biliary cells in a thin wall of fibrous tissue. Residual hepatocytes may be present beneath the fibrous layer.7
Differential Diagnosis
Isolated Polycystic liver disease (PLD)
As mentioned above, PLD is similar to ADPKD but without renal involvement. It is an autosomal dominant disease affecting 1 in 100,000 individuals. When identifiable, mutations involve the PRKSCH, SEC63, or LRP5 genes. The clinical course is less severe than ADPKD, and most patients are asymptomatic.8
Autosomal recessive polycystic kidney disease (ARPKD)
ARPKD is primarily a disease of the young and affects 1:20000 births, of which 30% may die from severe lung dysplasia and secondary respiratory failure, with renal collecting duct dilatation, bile duct dysplasia, and portal fibrosis. ARPKD is caused by a mutation in the PKHD1 gene, and the kidney and liver are always involved. Grossly, the liver in ARPKD appears normal to only slightly enlarged, and unlike ARPKD, extensive cysts formation is not seen. Microscopically, normal portal tracts containing a bile duct with a hepatic artery and a portal vein are severely diminished. There are persistent ductal plates arranged at the periportal areas and may show enlarged portal tracts with bridging fibrosis, which is indistinguishable from congenital hepatic fibrosis (CHF).9
Congenital hepatic fibrosis (CHF)
CHF is mostly seen in ARPKD patients, but it also occurs alone or is associated with Caroli disease (CD), juvenile nephronophthisis, Meckel-Gruber syndrome, ADPKD, and tuberous sclerosis.10 Many cases of CHF are autosomal recessive, and renal involvement is common. Microscopically, portal areas are expanded, with portal-portal bands of fibrosis and hypoplasia of portal veins. No regenerative hepatocytes are seen. The scarring shows irregular jigsaw patterns, which is typically seen in biliary diseases.7
Bile duct harmatoma (Von Meyenburg complex)
Bile duct hamartomas are irregular dilated cystic bile ducts, which are embryologic remnants that failed to involute during embryogenesis with unknown etiology. The prevalence is 0.6% to 5.6%, and it is predominantly incidental with low malignancy potential. It can also be a component with other entities such as ADPKD, CHF, or Caroli disease.7,11
Caroli disease (CD)
CD shows congenital dilatation of the larger intrahepatic bile. When there is duct dilatation in the setting of CHF, it is regarded as Caroli syndrome.9 There might be secondary infection and abscess formation in the dilated ducts. Microscopically, the biliary ductal epithelium is commonly normal, but it can be denuded, inflamed, or ulcerated. The differences among the above differential diagnosis are subtle since the histologic features are overlapping. Therefore, detailed clinical information is indispensable to render the correct diagnosis.
References
- de Miranda Henriques MS, de Morais Villar EJ. The Liver and Polycystic Kidney Disease. In: Li X, editor. Polycystic Kidney Disease [Internet]. Brisbane (AU): Codon Publications; 2015 Nov. Chapter 17. Available from: https://www.ncbi.nlm.nih.gov/books/NBK373392/ doi:10.15586/codon.pkd.20….
- Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med 1993;329:332-342.
- Cornec-LeGall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet 2019;393:919-935.
- Torres, V.E., Bennett, W.M. (2022). Autosomal dominant polycystic kidney disease (ADPKD) in adults: Epidemiology, clinical presentation, and diagnosis, UptoDate. Available from https://www.uptodate.com/contents/autosomal-dominant-polycystic-kidney-disease-adpkd-in-adults-epidemiology-clinical-presentation-and-diagnosis
- Cnossen WR, Drenth JP. Polycystic liver disease: an overview of pathogenesis, clinical manifestations and management. Orphanet J Rare Dis. 2014 May 1;9:69. doi: 10.1186/1750-1172-9-69.
- Zhang ZY, Wang ZM, Huang Y. Polycystic liver disease: Classification, diagnosis, treatment process, and clinical management. World J Hepatol. 2020 Mar 27;12(3):72-83. doi: 10.4254/wjh.v12.i3.72. PMID: 32231761; PMCID: PMC7097502.
- Lewis J. Pathology of Fibropolycystic Liver Diseases. Clin Liver Dis (Hoboken). 2021 May 1;17(4):238-243. doi: 10.1002/cld.1044. PMID: 33968382; PMCID: PMC8087910.
- Perugorria MJ, Masyuk TV, Marin JJ, et al. Polycystic liver diseases: advanced insights into the molecular mechanisms. Nat Rev Gastroenterol Hepatol 2014;11:750-761.
- Desmet V. Pathogenesis of ductal plate malformation. Mayo Clin Proc 1998;73:80-89.
- Guay-Woodford LM, Bissler JJ, Braun MC, Bockenhauer D, Cadnapaphornchai MA, Dell KM, Kerecuk L, Liebau MC, Alonso-Peclet MH, Shneider B, Emre S, Heller T, Kamath BM, Murray KF, Moise K, Eichenwald EE, Evans J, Keller RL, Wilkins-Haug L, Bergmann C, Gunay-Aygun M, Hooper SR, Hardy KK, Hartung EA, Streisand R, Perrone R, Moxey-Mims M. Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: report of an international conference. J Pediatr. 2014 Sep;165(3):611-7. doi: 10.1016/j.jpeds.2014.06.015. Epub 2014 Jul 9.
- Lanser HC, Puckett Y. Biliary Duct Hamartoma. [Updated 2022 Nov 23]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557750/