PATHOLOGY PEARLS: HEREDITARY HEMOCHROMATOSIS

Brief Case Presentation
The patient is a very pleasant 55-year-old male with no known past medical history. He began to have symptoms such as increased thirst, sweet taste in the mouth, fatigue, and unintentional weight loss in the past 6-8 weeks. Physical examination showed dark bronze skin and spider telangiectasia on the chest. The sclera was anicteric. No joint or abdominal tenderness. Due to markedly elevated HbA1c of 11, hyperglycemia, and ketonuria, he was admitted for further evaluation.
Lab
| Lab Test | Result | (Normal Range) | ||
| ALT | ↑ | 46 | (6-45 IU/L) | |
| AST | ↑ | 71 | (10-42 IU/L) | |
| Alkaline Phosphatase | 82 | (34-104 IU/L) | ||
| Total Bilirubin | ↑ | 1.6 | (0.2-1.3 mg/dl) | |
| Direct Bilirubin | 0.3 | (0.0-0.3 mg/dl) | ||
| Iron | 181 | (49-181 ug/dl) | ||
| Ferritin | ↑ | 2573 | (22-322 ng/ml) | |
| TIBC | ↓ | 217 | (250-450 ug/dl) | |
| Transferrin saturation | ↑ | 83% | (15-50%) |
An abdominal ultrasound showed an enlarged liver with a nodular surface. No intrahepatic biliary dilatation. There were small choleliths in the gallbladder without obstruction. The sonographic Murphy sign was negative.
A liver biopsy was subsequently performed.
Liver Core Biopsy Findings
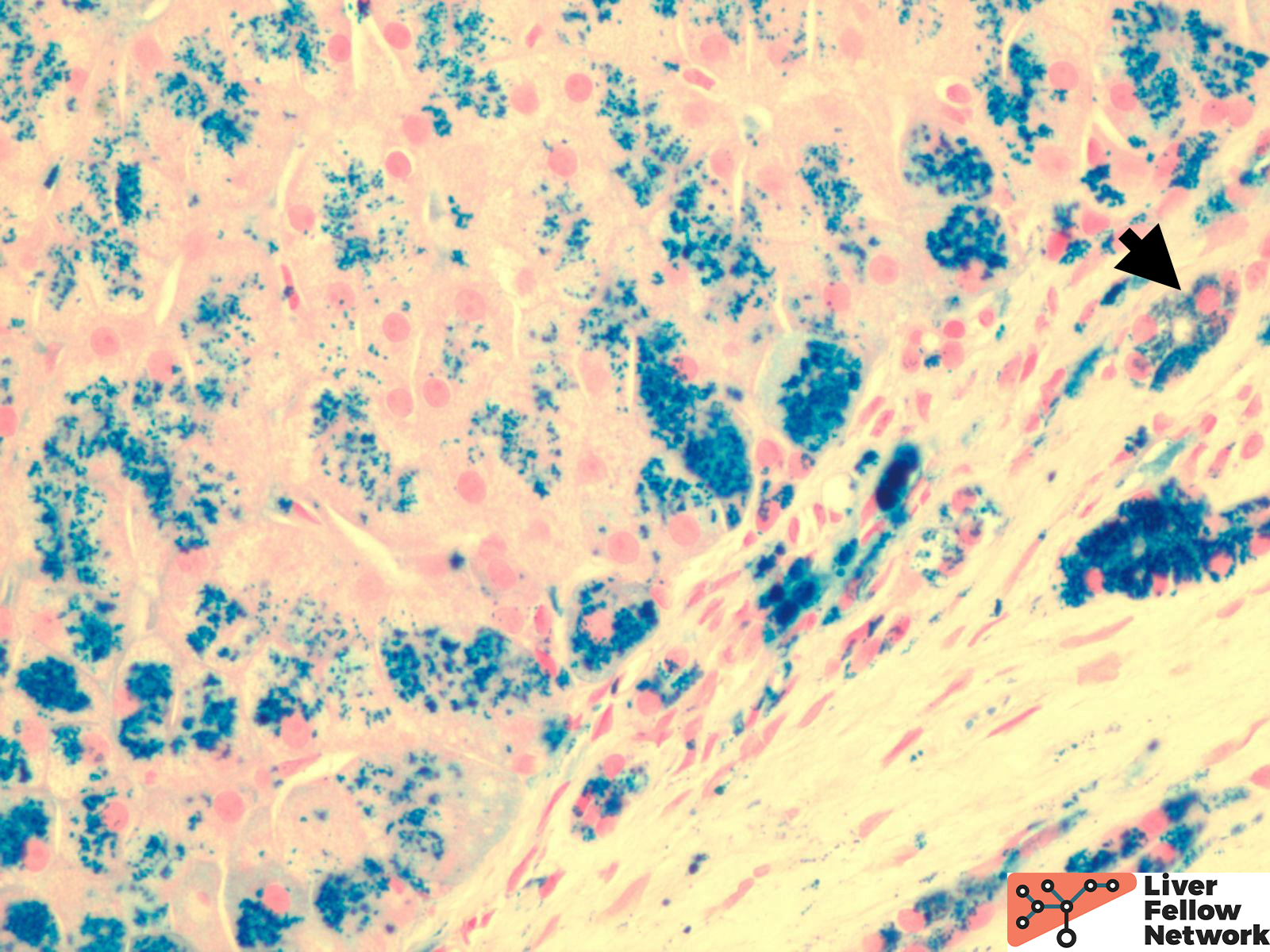
Low power shows minimal nonspecific mononuclear inflammation predominantly in the periportal area. There is mild fibrosis on H&E stain. There are numerous golden-brown fine pigment depositions in hepatocytes. (Figure 1,2)
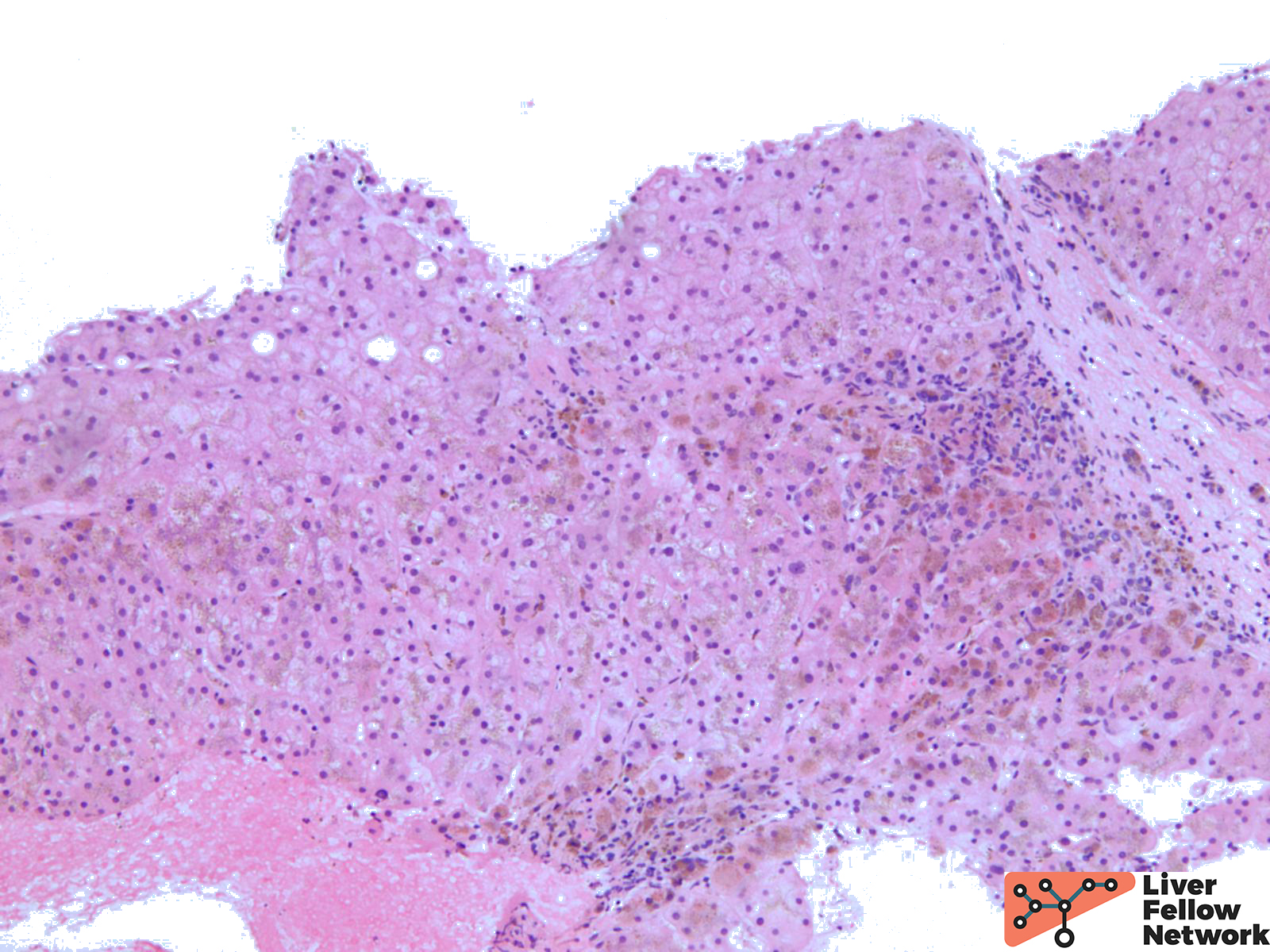
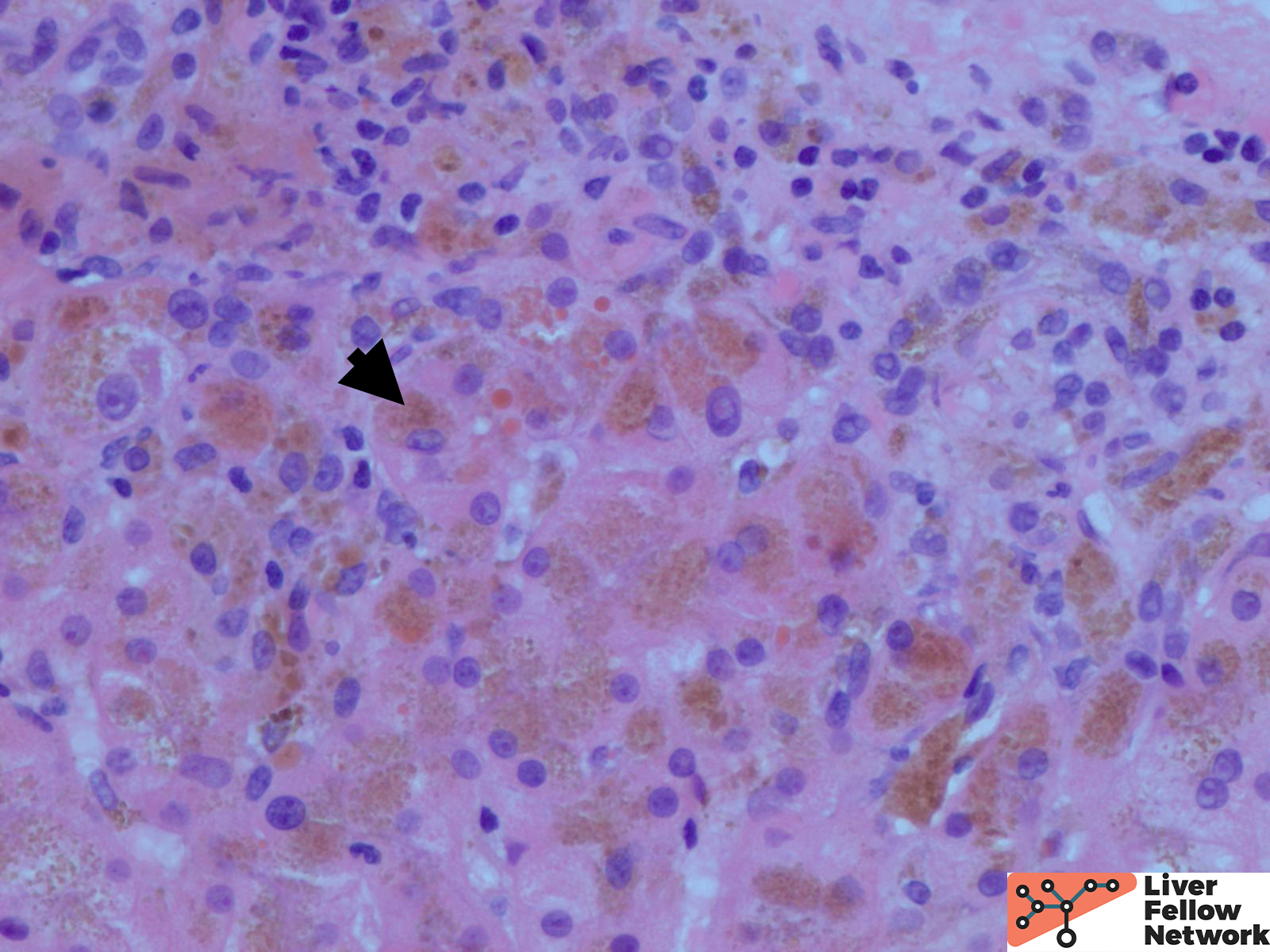
Iron stain highlights the dense iron deposits and confirms the golden-brown pigment is indeed iron. (Figure 3)
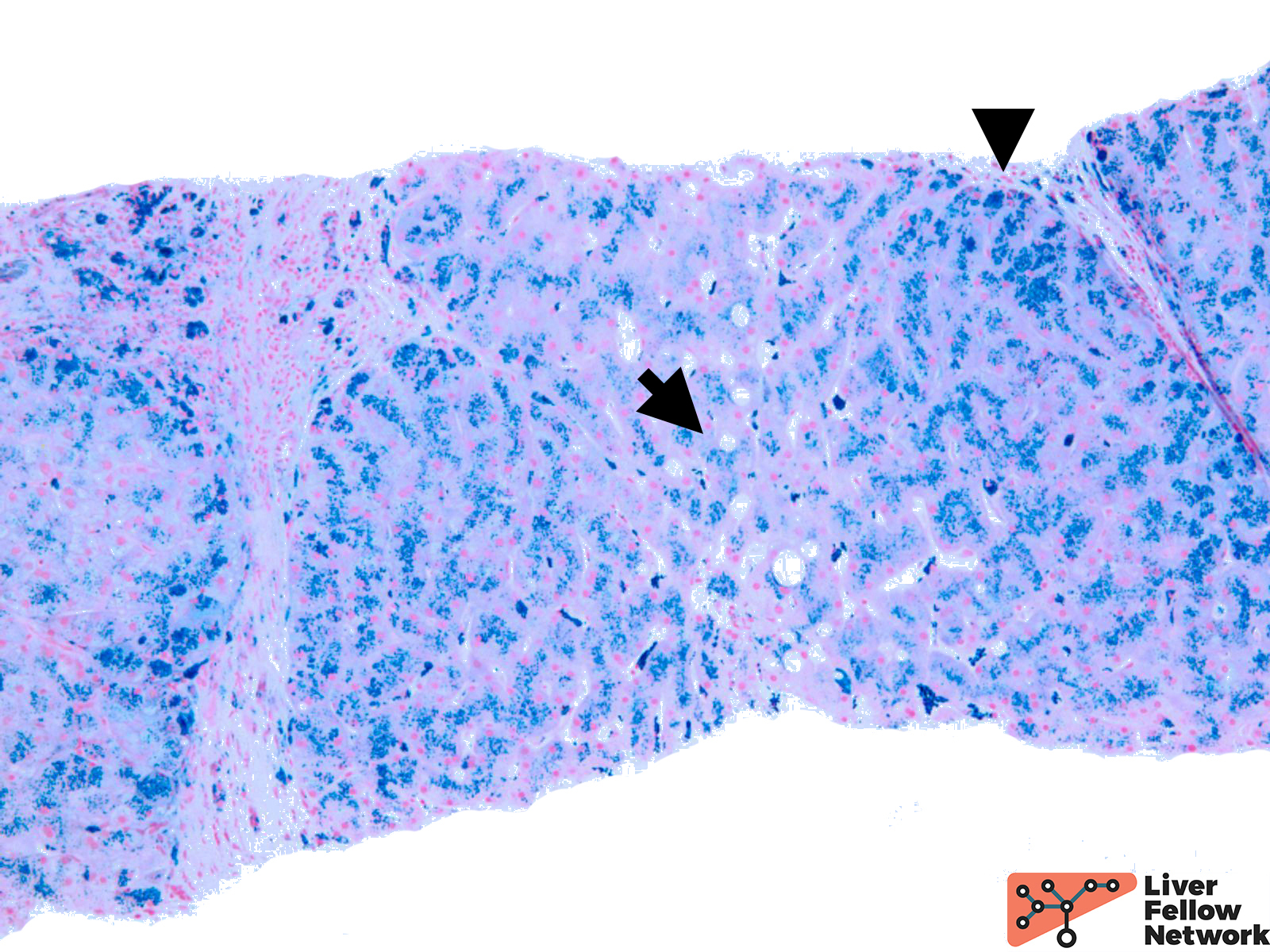
The biopsy findings are compatible with the diagnosis of iron overload. The patient underwent HFE mutation testing, which was positive for Homozygous c.845G>A, p.C282Y variant in the HFE gene.
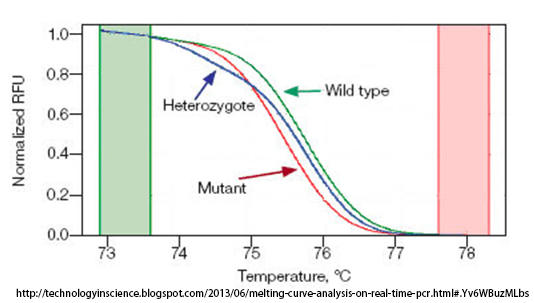
While Anatomic Pathologists examine tissue biopsy samples, such as the liver biopsy we have examined for this case presentation, genetic testing is performed in molecular biology laboratories and interpreted by a Clinical Pathologist. Detection of the most common variants that cause hemochromatosis are evaluated using polymerase chain reaction (PCR) and melting curve analysis. After isolating the patient’s DNA, allele-specific fluorescent probes are used to bind the patient’s DNA. These unique probes only release a signal when in double-stranded form (dsDNA). After the PCR reaction, the temperature of the PCR products is gradually raised until the double-stranded DNA (dsDNA) begins to break apart into single-stranded DNA (ssDNA), and the fluorescent signal is extinguished. If the fluorescent probes bind to wild-type DNA, they will do so tightly, whereas probes that bind to DNA containing a point mutation will bind with less affinity. Thus, the melting temperature of PCR products from a sample with the point mutation will be lower than from a wild-type control sample. The figure below shows an example melting curve showing the relative melting curves from wild-type, heterozygous mutant, and homozygous mutant sample.
The patient’s family history includes diabetes and hemochromatosis. Along with his high ferritin and transferrin saturation, the diagnosis was Hereditary Hemochromatosis.
Hereditary Hemochromatosis (HH)
Hemochromatosis is a disorder caused by excessive iron deposits in body tissues and organs such as heart, liver, pancreas, and joints, among others. Normally the iron level homeostasis is regulated in the human body through a balance of intake and excretion. Hemochromatosis occurs when there is pathological accumulation of iron. Hereditary hemochromatosis is an inherited autosomal recessive disorder characterized by increased iron absorption in the gut. Secondary hemochromatosis commonly occurs in the setting of long-standing blood transfusion, such as for the treatment for hematopoietic disorders. Over time, the destruction of red blood cells results in iron overload from heme products. Another fairly common setting for secondary hemochromatosis is dialysis and end-stage renal disease. Chronic liver diseases, parental iron overload, porphyria cutanea tarda, chronic hepatitis, alcoholic liver disease, or cirrhosis are also possible etiologies resulting in secondary hemochromatosis.
Hereditary Hemochromatosis (HH) is the most common genetic disorder in European ancestry with a prevalence of 1 in 300. It is extremely rare in individuals in East Asia (<0.1 percent). There are four types of hereditary hemochromatosis:
- Type 1 (HFE related) – Most common, adult-onset, 40-60 years. C282Y and H63D are the most common mutations of the HFE gene on chromosome 6.
- Type 2a (mutation of hemojuvelin gene, HJV), Type 2b (mutation of HAMP gene) – Early onset, 15-20 years.
- Type 3 (mutation of transferrin receptor- gene, TfR2) – Onset at 30-40 years.
- Type 4 (mutation of ferroportin gene) – Autosomal dominant, onset at 10-80 years, limited clinical consequences.
The typical lab profiles for HH are as follows.
- Elevated transferrin saturation (TSAT): The ratio of iron to total iron binding capacity (TIBC). Because TSAT is a ratio, it can be falsely elevated by conditions that acutely raise the serum iron level such as an episode of hemolysis or ingestion of an oral iron tablet.
- Elevated Ferritin: Ferritin is the circulating iron storage protein. It is made in the liver. Ferritin is also an acute phase reactant, so it can be elevated by chronic inflammation, acute liver injury, or other acute illnesses.
- Elevated transaminases: It is an indicator of liver injury, but they are not specific for HH.
- HFE mutation testing: The majority of HH is HFE related. However, there are other genes that are not covered by the testing panel.
With the advancement of imaging techniques combined with genetic testing, liver biopsy is no longer absolutely required for diagnosis in many cases. On the other hand, there are clinical scenarios such as equivocal imaging findings, rare genetic profiles other than common HFE mutations, and/or insufficient clinical evidence of HH; wherein liver biopsy remains key to establishing the correct diagnosis, especially of early or indolent cases of HH.
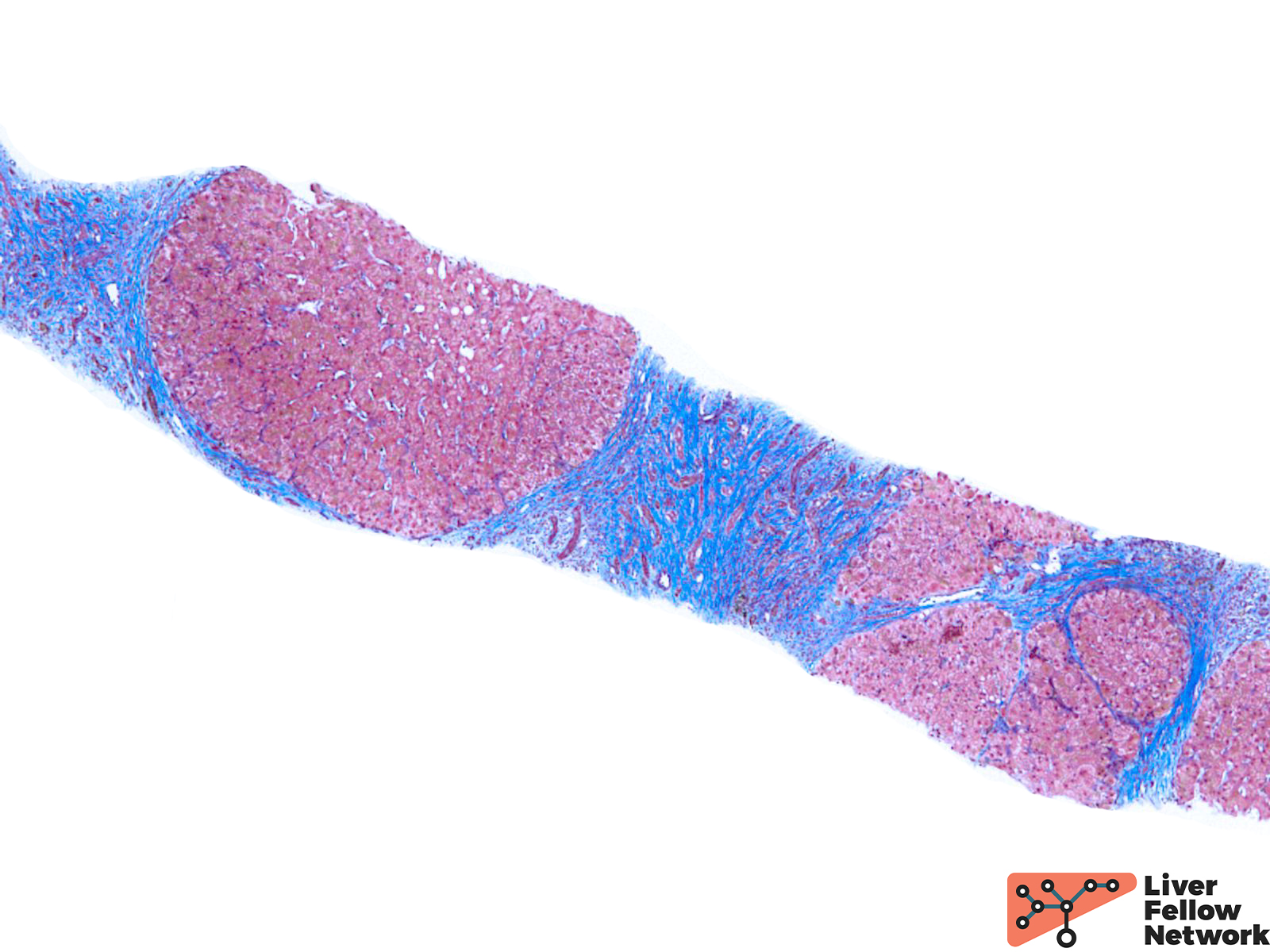
The liver biopsy plays an essential role in the follow-up management of HH patients; it is used to establish the liver fibrosis stage at the time of diagnosis. Timely and correct management may allow for regression of liver fibrosis and further reduce the risk of malignancy. Thus, liver biopsy is still indispensable to the management of HH.
Histologic Features of the liver in Hereditary Hemochromatosis
The patterns of iron deposition in HH may affect parenchymal cells (hepatocytes and bile duct cells) as well as mesenchymal cells (endothelial, Kupffer, and fat-storing cells from sinusoids). Its abundance and cellular distribution often differ from one area to another. There are three types of hepatic iron overload according to the cellular and lobular distribution of iron deposits:
- Parenchymal: Iron deposition within hepatocytes. Iron accumulates as fine granules mostly at the biliary pole and is distributed throughout the lobule with a decreasing gradient from periportal to centrilobular areas.
- Mesenchymal: At a later stage when hepatocytic iron is sufficiently high to induce cell necrosis. The iron deposition is within sinusoidal cells, mainly Kupffer cells and/or portal macrophages. The hepatocytic iron deposits are rough, sparse, and located close to iron-loaded macrophages.
- Mixed: Iron overload presents with the histologic characteristics as above two types.
Assessing the distribution patterns may give hints on the etiology of iron deposition disorders and predict the prognosis of the patients. During the evaluation, a predominant parenchymal iron deposition may give hints to an earlier stage in HH. With the disease progression, more mesenchymal patterns would be observed. (Figure 4) Meanwhile, the degree of iron overload is also assessed. There are a variety of grading schemes, but the Scheuer system is most commonly used. (Table 1) Lastly, fibrotic staging is critical to evaluating the overall prognosis and the risk of malignancy. (Figure 5)

Differential Diagnosis
While most HH is related to HFE gene, non-HFE mutation should be in the consideration if the HFE mutation test is negative and the clinical picture suspects HH. Apart from HH, non-genetic iron overload must be excluded. As mentioned above, there are numerous causes to iron overload. Here are some common differential diagnoses.
- Thalassemia
Transfusion can cause elevation of iron deposit, and ineffective erythropoietic drive also induces excess iron absorption from the dietary pool in patients with Thalassemia. The iron accumulates predominantly in the Kupffer cells and macrophages. - Sideroblastic anemia
The reason for the excessive iron overload in sideroblastic anemia is currently uncertain. The description of histological findings remains sparse, but the iron accumulation can be heavy, predominantly hepatocellular, and shows a zone 1 predominant pattern. - Porphyria cutanea tarda (PCT)
PCT is predominantly sporadic, while 20% of individuals will have a positive family history. It can be precipitated by hepatitis C virus (HCV) infection and alcohol use. Histological features vary from different predisposing factors such as alcohol and viral infection. Hepatic steatosis on liver biopsy is a near-universal finding. - Transfusion overload
Old RBCs are sequestered in Kupffer cells and macrophages. Histologically, iron is initially deposited in the zone 3 Kupffer cells and it gradually extends to the entire lobe. Fibrosis can develop with long-term transfusion-related iron overload. - Alcoholic liver disease
With increasing alcohol use, iron homeostasis is disturbed. Iron deposit is often mild and confined to periportal hepatocytes and Kupffer cells. - End stage cirrhosis
Iron overload may be seen in cirrhosis. The histological pattern is similar to HFE defects, as iron deposits predominantly in hepatocytes. However, the iron granules are typically more variable in amount from nodule to nodule.
Diagnosis of iron overload in the liver relies on thorough clinical radiological correlation with pathological examination. Molecular study also plays a key role in identifying genetic alterations in HH. Once the diagnosis is confirmed, a regular histological evaluation of disease progression is indispensable to optimize the management.
References
- Bacon, B.R., Phatak, P. (2022). Clinical manifestations and diagnosis of hereditary hemochromatosis. In T.W. Post, K. Lindor, J. Tirnauer & L. Kunins (Eds.), UptoDate. Available from https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-hereditary-hemochromatosis?search=Clinical%20manifestations%20and%20diagnosis%20of%20hereditary%20hemochromatosis&source=search_result&selectedTitle=1~120&usage_type=default&display_rank=1
- Pietrangelo, A. (2010). Hereditary hemochromatosis: Pathogenesis, diagnosis, and treatment. In Gastroenterology (Vol. 139, Issue 2). W.B. Saunders. https://doi.org/10.1053/j.gastro.2010.06.013
- Porter JL, Rawla P. Hemochromatosis. [Updated 2022 Jun 11]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK430862/
- Deugnier, Y., & Turlin, B. (2011). Pathology of hepatic iron overload. Seminars in Liver Disease, 31(3), 260–271. https://doi.org/10.1055/s-0031-1286057
- V. Bach, M.J. Barceló, A. Altés, et.al, Genotyping the HFE gene by melting point analysis with the LightCycler system: Pros and cons, Blood Cells, Molecules, and Diseases, Volume 36, Issue 2, 2006, Pages 288-291, https://doi.org/10.1016/j.bcmd.2005.12.030.
- Salomao MA. Pathology of Hepatic Iron Overload. Clin Liver Dis (Hoboken). 2021 May 1;17(4):232-237. doi: 10.1002/cld.1051.
- Burt, Alastair D, et al. MacSween’s Pathology of the Liver. 7th ed., Philadelphia Elsevier, 2018.
- Scheuer, P.J.; Williams, R.; Muir, A.R. Hepatic pathology in relatives of patients with haemochromatosis.J. Pathol. Bacteriol.1962,84, 53–64