Why does cirrhosis affect hemodynamics and cardiac function?

Why does cirrhosis affect hemodynamics and cardiac function?
Cirrhosis has a myriad of effects on human cardiovascular physiology. Many of the hemodynamic changes, pathophysiological processes, and complications that occur with cirrhosis can be traced back to the presence of portal hypertension. Furthermore, worsening portal hypertension correlates with increased morbidity and mortality. In this article from the Why? Series, we will explore the effects of cirrhosis and portal hypertension on hemodynamics as well as the pathogenesis and implications of cirrhotic cardiomyopathy.
Pathogenesis of the Hyperdynamic Circulation in Cirrhosis
Cirrhosis is defined by the presence of bridging fibrosis and regenerative nodules. As hepatic fibrosis progresses and other inflammatory and vasoactive processes ensue, resistance to blood flow in hepatic sinusoids increases. The resulting elevated portal venous pressure (portal hypertension) leads to shear stress on endothelial cells of the splanchnic vasculature, which produces nitric oxide (NO). In turn, NO causes further vasodilatation primarily in the splanchnic vasculature, as well as in the systemic vasculature via portosystemic collateral vessels.
Additionally, gut microbiota and endotoxemia also play major roles in the pathogenesis of portal hypertension, circulatory dysfunction, and hepatic decompensation. Vascular congestion within the splanchnic bed and subsequent intestinal edema lead to bacterial translocation from the gut into the splanchnic circulation and, through portosystemic collaterals, to the systemic circulation. Gut microbiota – particularly gram-negative organisms – trigger the release of pathogen-associated molecular patterns (PAMPs), lipopolysaccharides (LPS), and other pro-inflammatory cytokines and vasodilatory substances (such as TNF-α) that further augment splanchnic vasodilatation (Figure 1). In the liver, bacteria and inflammatory mediators damage sinusoidal endothelial cells (which are responsible for intrahepatic NO production) and activate hepatic stellate cells (HSCs). The combination of decreased intrahepatic NO production and HSC activation results in increased dynamic hepatic vascular resistance on top of the fixed resistance from underlying fibrosis, exacerbating portal hypertension. To summarize this contradictory role of NO in portal hypertension, there is a relative excess of NO in systemic circulation (splanchnic > peripheral) and a deficiency of NO in the liver itself.
Excessive exposure to vasodilatory mediators causes the splanchnic vasculature to enter a somewhat fixed vasodilated state, unable to robustly respond to vasoconstrictive stimuli. The peripheral circulation, on the other hand, is relatively less exposed to vasodilatory mediators and more able to vasoconstrict, partially offsetting the profound splanchnic vasodilatation to maintain mean arterial pressure (MAP). However, this is not without consequence. Because vasodilatation is more pronounced in the splanchnic circulation than in the peripheral circulation, blood flow preferentially enters the low-pressure splanchnic system. The result is low effective arterial blood volume and decreased perfusion of vital organs such as the kidneys and skeletal muscle, described as “splanchnic steal”. Renal hypoperfusion leads to sodium retention, free water retention, and expansion of total plasma volume (via renin-angiotensin-aldosterone (RAAS) and anti-diuretic hormone pathways) as well as cardiac output (CO) augmentation through sympathetic tone. This combination of low systemic vascular resistance (SVR), low MAP, and high CO is described as hyperdynamic circulation.
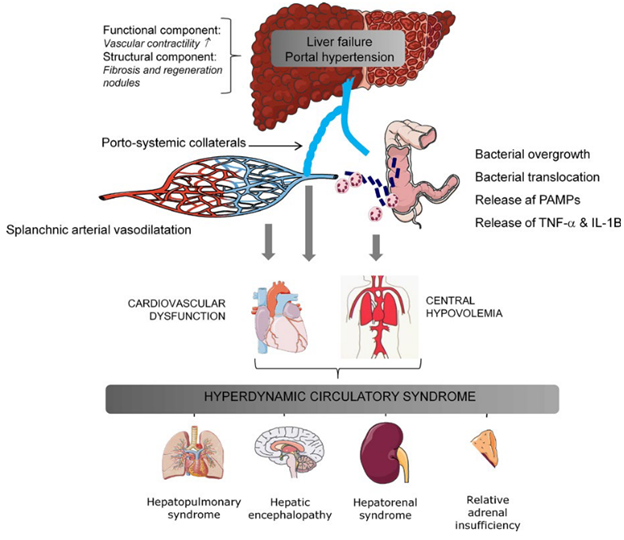
FIGURE 1. Illustration of mechanisms of development of the hyperdynamic syndrome in advanced cirrhosis and portal hypertension.
Møller S, Bendtsen F. The pathophysiology of arterial vasodilatation and hyperdynamic circulation in cirrhosis. Liver Int. 2018;38(4):570-580.
Targeting the Hyperdynamic Circulation
Hyperdynamic circulation is a consequence of portal hypertension, and it also becomes one of the driving forces responsible for progressing portal hypertension and decompensation. In essence, portal hypertension begets portal hypertension – ensuing a vicious cycle (Figure 2).
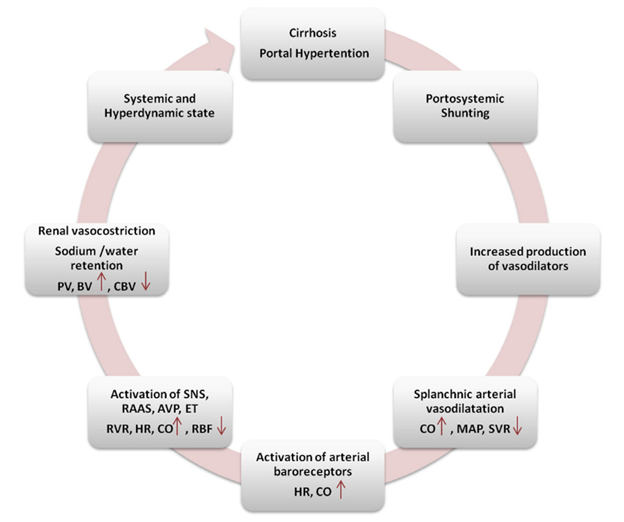
FIGURE 2. Mechanisms leading to hyperdynamic circulation.
Licata A, Mazzola A, Ingrassia D, Calvaruso V, Cammà C, Craxì A. Clinical implications of the hyperdynamic syndrome in cirrhosis. Eur J Intern Med. 2014;25(9):795-802.
For those reasons, hyperdynamic circulation is the target of several therapies used in the management of cirrhosis with portal hypertension. In a landmark trial on the use of non-selective beta-blockers (NSBBs) in compensated cirrhosis with portal hypertension, Villanueva et al. (2016) demonstrated that patients with clinically significant portal hypertension (CSPH) – defined as hepatic venous pressure gradient (HVPG) ≥ 10 mmHg – had more advanced hyperdynamic circulation at baseline and significantly higher HVPG reduction after intravenous β-blockade with propranolol than those with subclinical portal hypertension (HVPG < 10 mmHg). Furthermore, patients with CSPH and varices had more pronounced hyperdynamic circulation than those with CSPH and no varices. These findings supported the theory that hyperdynamic circulation is progressive and is more developed in higher degrees of portal hypertension.
Once the CSPH threshold has been crossed, hyperdynamic circulation (with increased CO) becomes a stronger exacerbator of portal hypertension due to a marked increase in portal venous inflow. These results were the impetus for the PREDESCI trial, which gave further credence to the use of NSBBs for prevention of decompensation in patients with CSPH and elevated carvedilol as the preferred NSBB in this setting. The PREDESCI trial demonstrated greater reductions in heart rate and cardiac index and improvement in HVPG in those treated with NSBBs (carvedilol > propranolol) compared to placebo – again illustrating the effect of NSBBs decreasing HVPG by attenuation of the increased portal inflow that occurs in hyperdynamic circulation. That all said, the benefits of beta-blockade in cirrhosis may be restricted to a specific window of opportunity. In later stages of portal hypertension, such as with refractory ascites, NSBBs’ blunting of cardiac output and sympathetic tone can contribute to renal hypoperfusion and exacerbate HRS-AKI. NSBBs should be used cautiously or perhaps avoided in these patients. For more on the use of NSBBs in cirrhosis, please refer to earlier posts in The Why? Series: Why do we use non-selective beta-blockers in cirrhosis? - Part 1 and Part 2.
Terlipressin, which is an analogue of lysine-vasopressin that acts on the V1 receptor, targets hyperdynamic circulation for the treatment of HRS-AKI via vasoconstriction predominantly in the splanchnic vascular bed. The hemodynamic effects of terlipressin are well-illustrated in a study using MRI after the first bolus of terlipressin in ten patients with HRS-AKI. After administration of terlipressin, subjects experienced redistribution of blood volume from splanchnic to systemic circulation via decreased splanchnic blood flow, decreased peripheral blood flow, and increased renal blood flow. Notably, increased renal blood flow with terlipressin occurs despite its negative effects on CO and heart rate. Reduction in HVPG with terlipressin is attributed more to its effects on splanchnic vasoconstriction than CO reduction.
Assessing Cardiac Function in Cirrhosis with Hyperdynamic Circulation
Accurately assessing cardiac function in the hyperdynamic state can be challenging. One may incorrectly assume that because both ejection fraction and CO are normal or increased, there must not be underlying cardiac dysfunction. However, low SVR in the hyperdynamic state has the potential to mask underlying cardiac dysfunction. Think of it this way: low SVR (and low MAP) in portal hypertension is somewhat analogous to aggressive afterload reduction, which is often intentionally pursued in the realm of cardiology for heart failure with reduced ejection fraction (HFrEF). Deficiencies in cardiac function may then become evident when confronted with stressors such as infection, GI bleeding, surgery, or TIPS. The term cirrhotic cardiomyopathy (CCM) was coined to describe this concept of adequate cardiac function at rest but inadequate response to physiologic stress in the cirrhotic patient.
Pathogenesis of Cirrhotic Cardiomyopathy
Briefly, CCM develops from the same pathophysiological processes that promote the development of the hyperdynamic state. Chronic sympathetic nervous system stimulation and cardiac β-adrenergic receptor activation eventually lead to downregulation of the cardiac β-adrenergic receptor and desensitization to sympathetic stimulation. Lee et al. (1990) demonstrated decreased density of cardiac β-adrenergic receptors and impaired myocardial response to catecholamine administration in cirrhotic rats compared to sham-operated controls. Harkening back to the influence of the microbiome and gut permeability, TNF-α and lipopolysaccharides in systemic circulation both negatively impact cardiac contractility. Impaired liver function and portosystemic shunting contribute to decreased clearance of and prolonged exposure to these vasodilatory and inflammatory mediators. Eventually, these processes lead to cardiac remodeling and/or decreased contractility which define CCM. Though there are other suspected mechanisms involved in the pathogenesis of CCM (Figure 3), this is an area of ongoing investigation. Lastly, the direct cardiotoxic effect of alcohol and the pathogenesis of alcohol-induced cardiomyopathy are also outside the scope of this review.
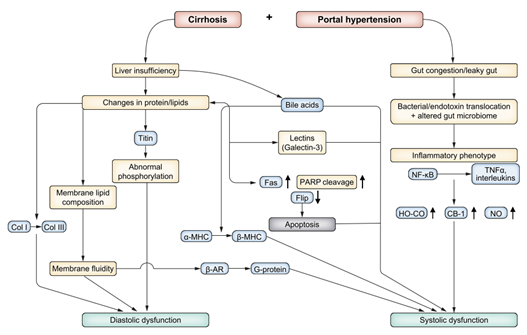
FIGURE 3. Mechanistic pathways of cirrhotic cardiomyopathy.
Liu H, Naser JA, Lin G, Lee SS. Cardiomyopathy in cirrhosis: From pathophysiology to clinical care. JHEP Reports. 2024-01-01 2024;6(1):100911.
Cirrhotic Cardiomyopathy Diagnostic Criteria
To give healthcare practitioners direction in diagnosing CCM, an expert panel at the Montreal 2005 World Congress of Gastroenterology proposed the initial diagnostic criteria (Table 1a). These criteria were a critically important step in unifying the field of hepatology to define, study, and better understand CCM. With time, however, multiple opportunities for improvement came to light. Firstly, the criterion “blunted contractile response on stress testing” had several issues. There is not a reproducible definition for it, which made our ability to reliably measure it in practice a concern. Additionally, many cirrhotic patients take non-selective beta blockers, which negatively affect inotropy. The criteria for diastolic dysfunction relied on parameters susceptible to influence by heart rate and loading conditions, which can vary significantly in patients with cirrhosis.

TABLE 1. Redefining Criteria for CCM.
Izzy M, Vanwagner LB, Lin G, et al. Redefining Cirrhotic Cardiomyopathy for the Modern Era. Hepatology. 2020;71(1):334-345.
Since the development of the WCG 2005 criteria, advancements in echocardiography – namely tissue doppler imaging (TDI) and speckle tracking echocardiography (STE) – improved our ability to recognize early signs of both systolic and diastolic dysfunction. While conventional Doppler imaging assesses the velocity of blood flow, TDI can track a focal point of myocardium and measure its velocity during a specified segment of the cardiac cycle. As a result of these echocardiography advancements, along with our evolving understandings and definitions of congestive heart failure, a multidisciplinary group of experts known as the Cirrhotic Cardiomyopathy Consortium met in Rochester, MN in October 2018 and subsequently proposed revised criteria for CCM in 2020 (Table 1b).
The 2020 criteria include several parameters that utilize TDI or STE: left ventricular global longitudinal strain (GLS), medial/septal e’ velocity (early diastolic medial mitral annular velocity), and E/e’ ratio (ratio of early diastolic mitral inflow velocity to early diastolic mitral annular velocity). GLS is a measure of the change in length of LV myocardium during systole relative to baseline length at diastole – in other words, shortening – and is thus represented as a negative percentage. A normal range of -18% to -22% was chosen for the 2020 CCM criteria because studies have observed both diminished and increased GLS in cirrhosis. Some studies have suggested that GLS increases (becomes more negative) as cirrhosis and portal hypertension advance. The heterogeneity of GLS in cirrhosis is likely due to multiple factors, including stage of cirrhosis, degree of systemic vasodilatation, comorbidities, and loading conditions, and warrants further investigation. The newer criteria’s septal e’ velocity by TDI – which is relatively preload-independent – more reliably measures myocardial relaxation and diastolic function. However, keep in mind that etiologies other than CCM (i.e., metabolic disease, advanced age, infiltrative disorders, etc.) can also cause systolic and diastolic dysfunction and the diagnosis of CCM mandates the absence of a known primary cardiac disease.
Estimating the prevalence of CCM has been challenging because diagnostic criteria have changed, screening for CCM is not routine, and volume status can fluctuate in cirrhotic patients. The 2005 CCM criteria likely overestimated the prevalence of CCM, in part due to sensitivity to volume status, and the prevalence of CCM using 2020 criteria is approximately 28-35%.
Outcomes Related to Circulatory Dysfunction and CCM
As mentioned earlier regarding the effects of beta-blockers and terlipressin on the hyperdynamic circulation, circulatory dysfunction is progressive and later stages are linked to negative outcomes. A prospective 2-year longitudinal follow up study using advanced cardiac imaging found indications of the temporal relationship between progressive cirrhosis and cardiac dysfunction. Specifically, increased left atrial volume index (via echocardiogram), abnormal myocardial extracellular volume (via cardiac MRI), and increased levels of natriuretic peptides were associated with death or need for liver transplant (LT). Patients who remained clinically stable throughout the study, on the other hand, did not demonstrate progressive cardiac dysfunction. Along similar lines, a prospective cohort study by Ruiz-del-Arbol and colleagues (2003) in patients with spontaneous bacterial peritonitis (SBP) found more pronounced baseline circulatory dysfunction in cirrhotic patients that later went on to develop HRS-AKI compared to those that did not that. These derangements included: lower SVR, lower MAP, lower stroke volume, lower CO, worse diastolic dysfunction, and more intense stimulation of RAAS and sympathetic nervous system
. Later research by the same group identified left ventricular diastolic dysfunction (LVDD) as an independent risk factor for HRS-AKI and mortality.
On the spectrum of circulatory dysfunction in cirrhosis, crossing the diagnostic threshold into CCM appears to confer additional risk for negative outcomes. Regarding renal outcomes, not only does CCM appear to increase the risk of developing HRS-AKI, but it may also negatively impact response to terlipressin according to a recent prospective study using point-of-care (POC) echocardiography. Relevant hemodynamic variables including E/e’ ratio, delta cardiac index, and NT-proBNP level were predictors of both terlipressin non-response as well as mortality. In another study using POC echocardiography in acute-on-chronic liver failure due to sepsis, the presence of CCM by 2020 criteria was an independent predictor of both circulatory failure and mortality when adjusted for age, gender, etiology of liver disease, obesity, diabetes, and MELD-Na. Finally, a retrospective cohort study of patients approved for LT at the Cleveland Clinic found that the presence of diastolic dysfunction per the 2020 CCM criteria was associated with increased pre-liver transplant mortality.
Development of Heart Failure after TIPS and Liver Transplant
Assessing CCM criteria and other parameters to estimate risk for development of new-onset heart failure following TIPS and LT is an area of active investigation. TIPS and LT are similar in that they resolve portal hypertension and significantly change resting hemodynamics; they clearly differ in that TIPS does not fix the liver dysfunction part of the equation. Placement of TIPS leads to a sudden increase in venous return to the heart, also known as preload, which can lead to heart failure in susceptible individuals. The incidence of post-TIPS heart failure is variable in the literature but appears to be approximately 10-20%. TIPS is generally contraindicated in the presence of HFrEF, so most attention on post-TIPS heart failure has been directed to diastolic dysfunction. Multiple studies have observed an association between pre-TIPS diastolic dysfunction and the development of heart failure post-TIPS. Increased E/e’ ratio and LA volume index, specifically, have been associated with post-TIPS HF. However, the data is mixed, as other studies have not observed an association with echocardiographic parameters and post-TIPS heart failure, but rather that right atrial pressure elevation pre-TIPS and immediately post-TIPS were predictive of heart failure.
Regarding post-LT heart failure, a retrospective analysis of 970 LT recipients at a single center in the United States over a 10-year period observed post-LT heart failure in 10.1%. Differentiating by HF subtype, the prevalences of post-LT heart failure reported in several studies range from 3-43% for HFpEF and 2-7% for HFrEF. The large spread for HFpEF is due to variability in the definition of HF and changes in the definition of diastolic dysfunction. As seen in the TIPS data, E/e’, LAVI, e’, and GLS have all been associated with higher risk of post-LT heart failure. A single-center retrospective cohort study of 166 patients observed higher pre-transplant H2FPEF scores in subjects that developed HF post-LT compared to those that did not develop HF post-LT; additionally, patients with MASH cirrhosis had higher H2FPEF scores than other etiologies of cirrhosis. Data on HF in the immediate post-LT setting are particularly limited, and uncertainty remains about the reversibility of CCM after liver transplantation.
There are a few limitations to our current understanding of post-TIPS and post-LT heart failure. Nearly all studies of heart failure after TIPS and LT are retrospective, and heart failure is a clinical diagnosis (see 2021 Universal Definition and Classification of Heart Failure). Accurate retrospective diagnosis of heart failure requires high-quality documentation and data. Furthermore, definitions of systolic, diastolic dysfunction, CCM, and advancements in echocardiography have changed over time, making it difficult to synthesize existing literature on the topic. Finally, indication for liver transplant has shifted dramatically over the last several decades as MASLD grows and hepatitis C diminishes, so results from older studies may not be as applicable to our modern patient population. To better understand heart failure after both TIPS and LT, larger, prospective, multicenter studies using standardized imaging protocols and consistent definitions of heart failure, systolic dysfunction, diastolic dysfunction, and CCM will be needed.
Increasing Prevalence of MASLD and Cardiac Dysfunction
In addition to the risk of developing cardiac dysfunction with CCM, patients with MASLD are also at risk for primary congestive heart failure – both HF with preserved ejection fraction (HFpEF) and HF with reduced ejection fraction (HFrEF). As MASLD increases in prevalence (estimated global prevalence ~30%) and thereby increases its share as etiology of cirrhosis for patients undergoing LT, so grows the focus on cardiac dysfunction in cirrhosis. The diagnosis of CCM requires the absence of a primary cardiac disorder, muddying the waters for understanding cardiac dysfunction in this population. Aside from the well-known impact of the individual metabolic conditions and the sum of conditions in metabolic syndrome on the risk for congestive heart failure, there is emerging evidence that MASLD is associated with cardiac remodeling that independently increases the risk of heart failure. Non-cirrhotic patients with MASLD carry a risk of heart failure, but the magnitude of the risk increases in parallel with the severity of liver fibrosis. Furthermore, the risk of heart failure in MASLD is tilted more toward HFpEF than HFrEF. Systemic insulin resistance is likely to contribute to this. Several phenotypes of HFpEF along the spectrum of MASLD to MASH cirrhosis have been proposed, and further studies are needed to better characterize this complex relationship and differentiation from CCM.
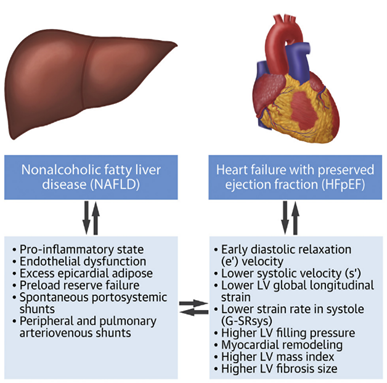
FIGURE 4. The Association Between Nonalcoholic Fatty Liver disease and Heart Failure with Preserved Ejection Fraction.
Salah HM, Pandey A, Soloveva A, et al. Relationship of Nonalcoholic Fatty Liver Disease and Heart Failure with Preserved Ejection Fraction. JACC Basic Transl Sci. 2021;6(11):918-932.
Conclusions
Cirrhosis with portal hypertension has profound effects on circulatory physiology. Hyperdynamic circulation becomes maladaptive, progressing portal hypertension and can lead to cardiac remodeling and dysfunction. However, due to low systemic vascular resistance, assessing cardiac function in cirrhosis is challenging as underlying cardiac dysfunction may be masked at rest. Criteria for CCM using echocardiography have been proposed to help clinicians recognize cirrhotic cardiomyopathy before it leads to decompensation. Further complicating the assessment of cardiac function and risk in cirrhosis is the growing prevalence of MASLD, which may independently increase the risk of heart failure. In this new era, cardiac assessment in cirrhosis requires a holistic and multidisciplinary approach to increase the recognition of these conditions, develop new treatments to modulate the trajectory of circulatory dysfunction, and decrease the risk of negative outcomes in this patient population.